

5.1. Сочетание масс-спектрометрии и хроматографии для определения аминокислотного состава белка
Методика анализа включает следующие операции.
К 50–100 мкг белка добавляют 16 меченных (дейтерированных) 18O амино-кислот в качестве внутреннего стандарта и высококонцентрированную соляную кислоту, содержащую 0,5–1,0 мас.% фенола, смесь выдерживают в вакууме при температуре t = 100ºС, упаривают и высушивают. Затем к остатку добавляют н-бутанол, воздействуют ультразвуком в течение 1 мин, запаивают в ампулу и выдерживают в масляной бане. После вскрытия ампулы проводят обработку смеси ангидридом трифторуксусной кислоты. Хроматографирование проводят на спиральной колонке, заполненной хромосорбом W.
В качестве примера на рис. 5.2 приведена хроматограмма, полученная при разделении гидролизата инсулина.
Сигнал
1 2 3 4 5 6 Время, мин

Рис. 5.2. Пики н-бутиловых эфиров аминокислот: 1 – аланин; 2 – треонин;
3 – глицин; 4 – валин; 5 – лейцин; 6 – пролин
В ХХ столетии были сняты полные масс-спектры всех компонентов и по характерному для каждой аминокислоты фрагменту качественно определены изотопные отношения. Для количественного анализа использовали набор внут-ренних стандартов – меченых аминокислот.
Меченые аминокислоты могут быть получены по способу Мерфи. Он за-ключается в нагревании при 60–70 ºС 1–4 мг той или иной аминокислоты с 0,1–0,2 мл подкисленной Н218О в течение нескольких суток. Включение метки со-ставляет 90% [5].
6. Полярографический метод
Фундаментальные основы метода были заложены Гейровским в 1922 г.
Установка для полярографического анализа представлена на рис. 6.1.
В электролизер 3 помещают анализируемый раствор 2. Через электролизер протекает ток с непостоянным напряжением, что достигается за счет делителя напряжения 7 и фиксируется с помощью гальванометра 6.
Анодом служит слой ртути 1 на дне ячейки, а в качестве катода выступает ртутный капельный электрод 4, через который проходит ртуть из резервуара 5.
Если повышать напряжение до достижения потенциала восстановления, ионы анализируемого вещества разряжаются на ртутном катоде, что приводит в итоге к возрастанию силы тока (I) в цепи.
Поскольку к катоду непрерывно доставляются новые количества ионов, даже в условиях снижения концентрации ионов у поверхности ртутного катода, сила тока не падает, а принимает постоянное значение, не зависящее от напряжения. Это явление реализуется при условии, что скорость катодного вос-становления равна скорости диффузии.
Зависимость силы тока (I) от приложенного напряжения (Е) представлена на рис. 6.2.
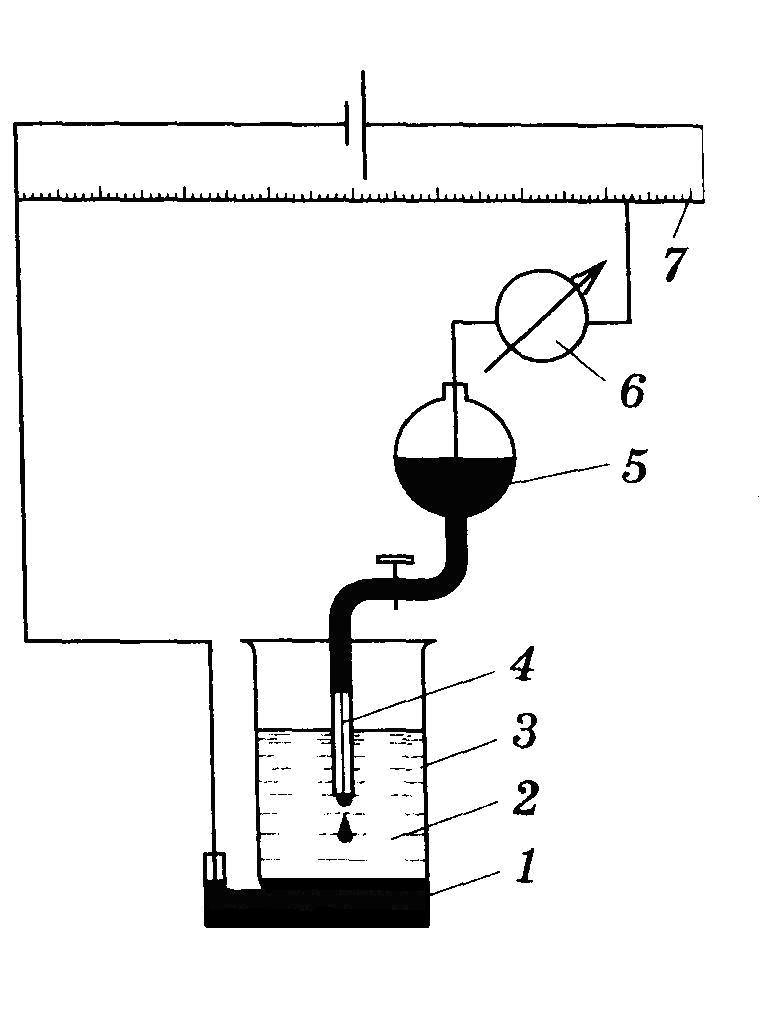
Рис. 6.1. Схема установки для полярографии:
1 – слой ртути; 2 – анализируемый раствор; 3 – электролизер; 4 – ртутный капающий электрод; 5 – резервуар ртути; 6 – гальванометр; 7 – реохорд (делитель напряжения)
Как видно из рис. 6.2, остаточный ток (0,1 мкА) реализуется при относительно небольшом потенциале катода, а подавление избыточного миграционного тока достигается введением в анализируемый раствор так назы-ваемого индифферентного электролита, имеющего более отрицательный по-тенциал выделения и экранирующего электрод своими катионами.
Зависимость I от E при обратимом электродном процессе, в соответствии с уравнением полярографической волны, подчиняется выражению:
E
= E1/2![]() ,
(6.1)
,
(6.1)
где E1/2 – потенциал полуволны;
Id – диффузионный ток.
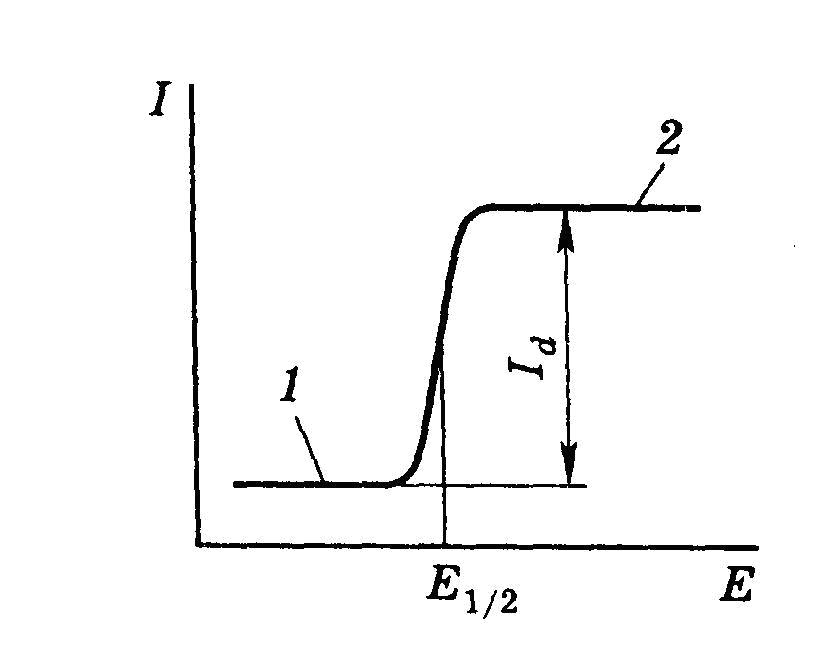
Рис. 6.2. Общий вид полярограммы при анализе пищевых продуктов: 1 – остаточный ток; 2 – диффузионный ток
Для необратимых процессов уравнение (6.1) имеет более сложную форму, однако при условии I = 1/2 Id:
E = E1/2 , (6.2)
и,
таким образом, потенциал полуволны не
зависит от тока и E1/2
![]() f
(c),
где c
–
концентрация восстановленного иона.
f
(c),
где c
–
концентрация восстановленного иона.
Поэтому определение потенциала полуволны лежит в основе проведения качественного анализа в полярографии. Если в растворе находится n веществ, способных к восстановлению на ртутном катоде, полярограмма будет содер-жать n «ступеней» (см. рис. 6.3).
По измеренному потенциалу полуволны E1/2 можно идентифицировать не-известный ион относительно того или иного фонового электролита.
В основе количественного анализа лежит уравнение, связывающее диффу-зионный ток (Id) с концентрацией иона (c), коэффициентом диффузии (D), массой ртути (m), вытекающей из капилляра за 1 с, и временем образования капли (τ):
Id = 605 z D1/2 m3/2 τ1/6 с , (6.3)
где z – заряд иона.
С учетом постоянных условий полярографирования, z, D, m и τ – величины постоянные, и тогда уравнение (6.3) приобретает вид:
Id = κ с. (6.4)
В соответствии с выражением (6.4) градуировочный график в координатах высота полярографической волны (Id) – концентрация восстановленного иона (с) представляет собой прямую линию, проходящую через начало координат.
График строится по стандартным растворам в количестве n ≥ 3. Однако следует отметить, что точность метода высока лишь в том случае, когда усло-
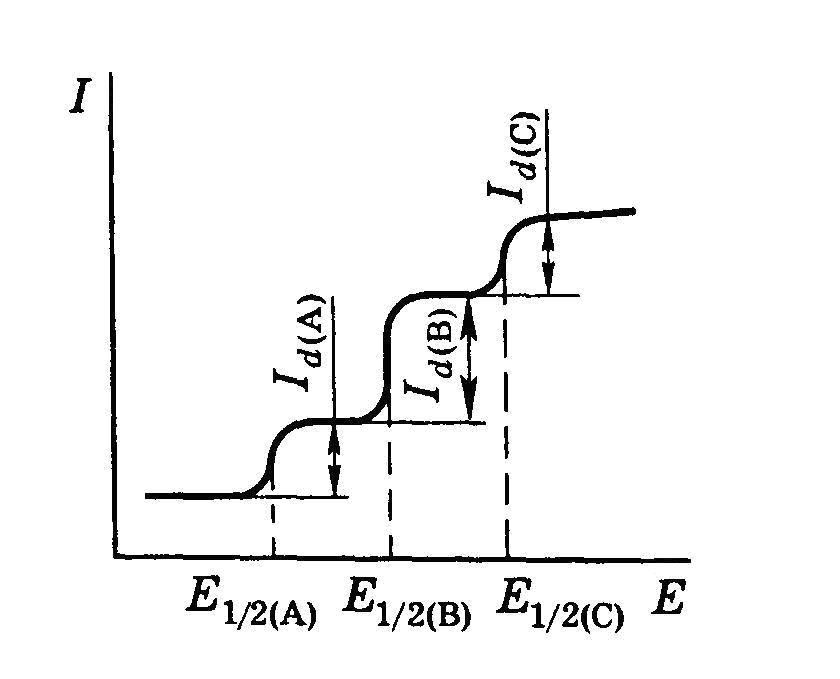
Рис. 6.3. Вид полярограммы при наличии в растворе трех восстанавливающихся веществ А,В и С: А – Cd; B – Ni; С – Zn
вия полярографирования для стандартных растворов и анализируемой пробы абсолютно идентичны, т.е. соблюдается равенство:
![]() .
(6.5)
.
(6.5)