

Таблетки палиперидона пролонгированного действия в профилактике рецидивов симптомов у больных шизофренией. Рандомизированное двойное слепое плацебо−контролируемое клиническое исследование (расширенный реферат)
М.Kramer, G.Simpson, V.Maciulis, S.Kushner, U.Vijapurkar, P.Lim, M.Eerdekens
США, Литва
Палиперидон является основным активным метаболитом хорошо известного антипсихотика рисперидона, действует на моноаминергические рецепторы аналогично рисперидону и другим новым антипсихотикам. Палиперидон пролонгированного действия (ER) – особая таблетированная форма, созданная на основе технологии OROS (корпорация "Alza", Маунтин-Вью, штат Калифорния, США) для непрерывного длительного высвобождения активного вещества в течение более 24 ч, при примении которой не происходит изменения его концентрации в плазме, характерного для препаратов с немедленным высвобождением. Профиль постепенного высвобождения палиперидона ER позволяет начинать лечение с эффективной терапевтической дозы без предварительного титрования. Период полувыведения палиперидона составляет около 1 сут вне зависимости от формы выпуска препарата.
Палиперидон ER в отличие от рисперидона не подвергается метаболизму в печени, что снижает риск клинически значимого взаимодействия с другими лекарственными препаратами. Это существенное преимущество для больных шизофренией, часто страдающих сопутствующими заболеваниями, которые требуют дополнительного медикаментозного лечения.
Эффективность и безопасность палиперидона ER при лечении обострений шизофрении продемонстрированы в трех завершенных рандомизированных двойных слепых контролируемых клинических исследованиях, в которых получены сопоставимые положительные результаты. Целью данного клинического исследования являлась оценка эффективности и безопасности палиперидона ER в профилактике рецидивов у пациентов, стабилизированных после активного эпизода шизофрении.
Материалы и методы
Пациенты
В исследование были включены мужчины и женщины в возрасте от 18 до 65 лет, у которых: 1) диагностирована шизофрения (Руководство по диагностике и статистике психических заболеваний, 4-е издание [DSM-IV]) в течение минимум 1 года и 2), имелся активный эпизод шизофрении (общий балл по шкале оценки позитивных и негативных синдромов – PANSS – 70–120). Кроме того, пациенты были физически здоровы, способны самостоятельно принимать назначенные препараты или имели помощника на протяжении всего исследования, а также могли самостоятельно заполнять опросники.
Пациентов не включали в исследование, если у них были выявлены другие заболевания I оси DSM-IV (не шизофрении), злоупотребление алкоголем или наркотиками согласно I оси DSM-IV (кроме никотина или кофеина) в течение 6 мес до скрининга, а также при высоком риске суицидального или агрессивного поведения. Пациентов исключали из исследования при наличии заболеваний, способных влиять на всасывание, метаболизм или выведение исследуемого препарата; тяжелого или нестабильного заболевания в анамнезе; аллергической реакции на барбитураты, карбамазепин, ламотриджин, фенитоин, палиперидон или рисперидон в анамнезе; в случае неэффективности лечения рисперидоном в анамнезе; применения антипсихотических препаратов пролонгированного действия в течение предыдущих 120 сут; применения исследуемого препарата в течение 90 сут, предшествующих скринингу; электросудорожной терапии в течение 3 мес, предшествующих скринингу, или принудительной госпитализации в психиатрическую больницу. Женщины не включались в случаях беременности – имеющейся или запланированной, – а также при кормлении грудью. Пациентов просили воздерживаться от употребления алкоголя и наркотиков в течение всего исследования.
До вводного периода отменяли противопаркинсонические и противоэпилептические препараты, препараты лития, β -адреноблокаторы (кроме случаев лечения артериальной гипертонии у стабильных пациентов) и ингибиторы моноаминоксидазы. Разрешено было применять пероральные бензодиазепины. После исходного визита для устранения экстрапирамидных нарушений разрешено было применять пероральные формы бензтропина или биперидина (или аналогичные препараты), а также β -адреноблокаторы для лечения обусловленной терапией акатизии. Антидепрессанты (кроме ингибиторов моноаминоксидазы) разрешалось применять, если в течение как минимум 3 мес до скрининга доза оставалась стабильной. Также применялась поддерживающая или обучающая психотерапия.
Дизайн исследования
Данное клиническое исследование, профинансированное компанией "Johnson & Johnson Pharmaceutical", проводили с 13 апреля по 31 августа 2005 г. в 45 центрах 6 стран (США, Румыния, Турция, Латвия, Литва, Индия). Дизайн исследования включал 5 этапов: скрининг, 8-недельный вводный период, в течение которого пациентов госпитализировали и открытым методом назначали палиперидон ER (3–15 мг один раз в сутки, начальная доза – 9 мг) до стабилизации состояния (минимум 2 нед), 6-недельная открытая фаза стабилизации, в ходе которой выписанные из стационара пациенты продолжали получать ранее подобранную дозу, двойная слепая фаза лечения разной продолжительности, в ходе которой стабильные пациенты были рандомизированы в соотношении 1:1 (с помощью компьютерной схемы рандомизации и стратификации с использованием голосовой интерактивной системы IVRS, предоставленных спонсором) в группы палиперидона ER (начиная с дозы, применяемой в период стабилизации) или плацебо, и возможное 52-недельное открытое продолжение исследования. Пациенты находились в двойной слепой фазе до появления развития рецидива, до выбывания из исследования или до завершения исследования. В данной статье представлены результаты, полученные на момент окончания двойной слепой фазы.
Независимым комитетом по мониторингу данных проводился постоянный мониторинг безопасности, оценивалась эффективность при промежуточном анализе и формулировались рекомендации спонсору относительно изменений, прекращения или продолжения исследования.
Показатели эффективности
Первичным показателем эффективности являлось время до первого рецидива в ходе двойной слепой фазы. Рецидив определялся при наличии одного из следующих критериев: 1) госпитализация (принудительная или добровольная) в психиатрический стационар; 2) увеличение общего балла PANSS на 25% в течение 2 сут подряд у пациентов с общим баллом более 40 на момент рандомизации или увеличение общего балла на 10 у пациентов с общим баллом до 40 на момент рандомизации; 3) увеличение балла по шкале общего клинического впечатления – тяжести симптомов (CGI-S) минимум до 4 у пациентов с баллом по этой шкале, равным 3 и менее на момент рандомизации, или минимум до 5 в течение 2 сут подряд у пациентов с баллом, равным 4 на момент рандомизации; 4) умышленное нанесение себе вреда или агрессивное поведение, или мысли о суициде или убийстве и клинически значимое агрессивное поведение; 5) увеличение баллов по отдельным пунктам PANSS минимум до 5 у пациентов с числом баллом 3 и менее на момент рандомизации или до 6 у пациентов с баллом 4 на момент рандомизации в течение 2 сут подряд.
Показатели безопасности и переносимости
Безопасность анализировали, оценивая нежелательные явления, обусловленные лечением (с использованием терминологии нежелательных явлений ВОЗ), клинические лабораторные показатели, основные физиологические показатели, массу тела, а также данные общего осмотра, данные электрокардиограммы в 12 отведениях и шкалы оценки двигательных нарушений (шкала Симпсона–Ангуса, шкала акатизии Барнса, шкала оценки аномальных непроизвольных движений). Также измеряли уровень пролактина в сыворотке.
Статистический анализ
Данное исследование включало заранее запланированный промежуточный анализ, проведенный независимым комитетом по мониторингу данных после 43 случаев обострений. При выявлении статистически значимой эффективности ( p =0,01) клиническое исследование прекращали.
Исследование имело 90% статистическую мощность для выявления 20% различия между 10-месячной частотой обострений на плацебо (45%) и палиперидоне ER (25%) или, равноценно, относительных риск, равный 0,48, при уровне статистической значимости 0,05. Существовала примерно 43% вероятность преждевременного прекращения исследования на основе достижения эффективности при промежуточном анализе.
Основной показатель эффективности (время до рецидива) анализировали методом Каплана–Мейера для оценки интегральной функции распределения. Для суммирования времени до рецидива по числу эпизодов, числу цензурированных пациентов, медиане, 25-му и 75-му процентилям эпизодов, а также для сравнения различий между разными препаратами применяли двусторонний лог-ранговый критерий. Расчет относительного риска и доверительного интервала (ДИ) для 95% вероятности проводили на основе модели Кокса для пропорциональных рисков, где единственной независимой переменной был тип применяемого препарата.
Все дополнительные анализы проводили при уровне статистической значимости, равной 0,05 (двусторонний тест), по группам, в которых применяли разные препараты, без поправки на множественность. Для анализов данных, полученных в ходе двойного закрытого этапа, применяли анализ с использованием последнего наблюдения (LOCF).
Анализируемые популяции пациентов
Все анализы эффективности по данным двойной слепой фазы включали пациентов популяции "intent-totreat", куда вошли все пациенты, прошедшие рандомизацию и получившие хотя бы одну дозу препарата, назначенного двойным слепым методом, и имевшие как минимум одну оценку эффективности после исходного срока.
Р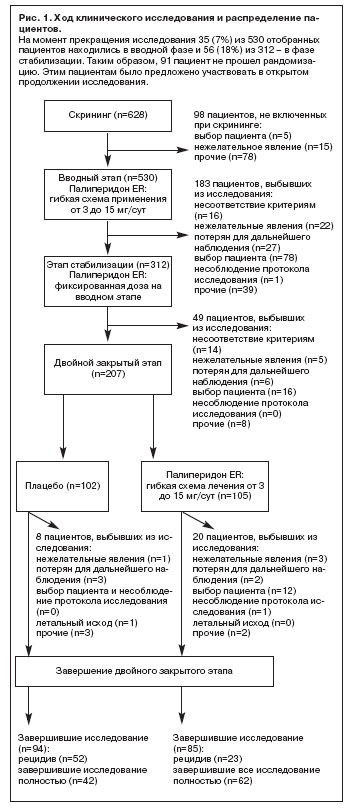 езультаты
езультаты
Решение о досрочном прекращении данного клинического исследования приняли на основе статистически значимых результатов по эффективности согласно определению Независимого комитета по мониторингу данных 1 августа 2005 г. Основным согласно протоколу был заранее заданный промежуточный анализ. Набор данных для подтверждающего окончательного анализа включил эпизоды, отмеченные с 13 мая 2005 г. (точка отсечения для промежуточного анализа) по 31 августа 2005 г.
Распределение и популяции пациентов
У 2 из 113 пациентов, прошедших рандомизацию, в момент отсечения при анализе результатов промежуточного анализа отсутствовали данные по эффективности, поэтому в популяцию intent-to-treat вошли 111 пациентов.
Пациенты, участвовавшие в клиническом исследовании в момент его досрочного прекращения, отнесены к категории завершивших исследование: 530 пациентов были включены на вводном этапе и получили как минимум одну дозу исследуемого препарата; 207 пациентов прошли рандомизацию для участия в двойной слепой фазе (рис. 1) . Из 179 пациентов, завершивших исследование, у 75 (36%) развился рецидив, а 104 (50%) пациента были отнесены к категории завершивших исследование, поскольку в момент прекращения клинического испытания они находились в двойной слепой фазе.
Случаи выбывания из исследования в двойной слепой фазе по разным причинам, кроме развития рецидива, отмечались чаще среди пациентов, получавших палиперидон ER (n=20; 19%), чем плацебо (n=8; 8%). Основной причиной исключения из группы палиперидона ER был отзыв согласия на участие в исследовании (n=12; 11%). В группе, получавшей плацебо, согласие на участие исследовании не отозвал ни один пациент.