

Коррозия с кислородной деполяризацией, основные особенности, влияние различных факторов.
Кислородная деполяризация – это коррозия под действием растворённого в коррозионной среде кислорода.
1) В кислых средах: О2+4Н++4ё=>2Н2О 2) В нейтральных и щелочных: О2+2Н2О+4ё=>4ОН-
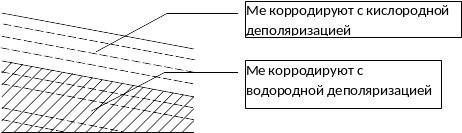
При коррозии с кислородной деполяризацией корродируют все Ме, кроме золота.
Стадии:
Доставка О2 – замедлена чаще всего.
Стадия разряда – замедленна, если быстро идёт доставка(в сильно перемешивающихся электролитах и в очень тонких плёнках влаги).
Отвод Н2О или Н3О+ не может быть замедлен.
Если замедлен разряд, то на коррозию с кислородной деполяризацией влияют те же факторы, что и на водородную деполяризацию(температура, природа Ме, рН, ПАВ, присутствие посторонних ионов и примесей).
Важная особенность кислородной деполяризации – очень частое возникновение аэрационных пар (пара зон: катодная и анодная):
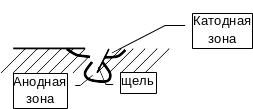 Кислороду легче
продиффундировать к катодным зонам, в
анодные труднее. Аэрационные пары
возникают из-за неодинаковой диффузии
кислорода к различным участкам, это
приводит к локализации коррозионного
процесса. Анодная зона локализуется
на участке, где более затруднена диффузия
кислорода. Анодный процесс локализуется
в глубине трещины и трещина развивается
вглубь – эффект щелевой коррозии.
Кислороду легче
продиффундировать к катодным зонам, в
анодные труднее. Аэрационные пары
возникают из-за неодинаковой диффузии
кислорода к различным участкам, это
приводит к локализации коррозионного
процесса. Анодная зона локализуется
на участке, где более затруднена диффузия
кислорода. Анодный процесс локализуется
в глубине трещины и трещина развивается
вглубь – эффект щелевой коррозии.
Коррозия металлов в контакте, основные особенности, использование в практике защиты.
При контакте двух Ме на поверхности обоих Ме устанавливается средний потенциал коррозии, при этом потенциал коррозии более отрицательного Ме сдвигается в положительную сторону и скорость его коррозии резко увеличивается, а потенциал более положительного Ме сдвигается в отрицательном направлении – скорость коррозии уменьшается или прекращается полностью.
Особенности контактной коррозии:
Коррозионный процесс локализуется на более эл. отрицательном Ме и конструкция выходит из строя быстрее, поэтому существует ГОСТ ЕСЗКС 9.005-74 «Допустимые и недопустимые контакты Ме». Он даёт допустимые «+» и недопустимые «-« контакты Ме. Если контакт недопустим «-«, то применять его можно только при проведении специальных мероприятий по предотвращению контактной коррозии.
Контактная коррозия зависит от эл.проводности коррозионной среды.
В контакте коррозия более электроположительного Ме уменьшается или прекращается совсем – это свойство применяется на практике в качестве протекторной защиты, т.е. присоединение к конструкции анодного протектора Ме (Zn, Mg или их сплавов), электродный потенциал и поверхность которого обеспечивают катодную поляризацию всех остальных Ме конструкции, т.е. перевод их в катоды.
![]()







Пассивное состояние металлов. Современная теория пассивности. Использование явления пассивности в практике защиты.
Пассивность – это состояние повышенной коррозионной стойкости Ме, вызванное торможением анодного процесса за счёт образования упорядоченных плотных оксидных, гидроксидных адсорбционных и солевых плёнок. Первым это явление наблюдал Ломоносов М.В.(Fe перестаёт корродировать в HNO3, при её высокой концентрации).\
Виды пассивации: 1) Солевая; 2) Вызванная образованием оксидов, гидроксидов (кислородсодержащих соединений). Задаём потенциал и фиксируем ток:
Влияние СL- на поляризационную кривую
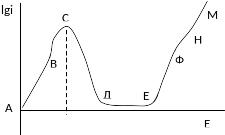

Теории пассивности:
Фазовая теория пассивации – пассивность вызывают оксиды и гидроксиды.
Участок АВ: Ме – nё => Ме2+.
Точка В – образование оксида, потенциал начала пассивации: ииМе – nё + Н2О => МеО + 2Н+.
Точка С – потенциал начала видимой пассивации, резкое снижение i.
Точка Д – потенциал полной пассивации (потенциал активации, если снимаем с положительного потенциала) или потенциал фладе.
Участок ДЕ – участок полной пассивации, идут одновременно два процесса: МеО + 2Н+ => Ме2+ + Н2О – растворение оксида, скорость определяющая реакция;
Ме – nё + Н2О => МеО + 2Н+ - возобновление оксида под действием эл.химической реакции. Скорость химической реакции не зависит от потенциала.
Точка Е – начинается окисление Ме до более высоких степеней окисления: Ме – 3ё + 2Н2О => МеО2- + 4Н+ - потенциал перепассивации.
Участок ФН – предельный ток по отводу сложных анионов.
Точка Н – выделение кислорода: Н2О – 4ё => О2 + 4Н+.
Если Ме выделить при ЕПАССИВ, то на поверхности Ме можно обнаружить оксид или гидроксид, но теория оксида не могла объяснить всю совокупность экспериментальных данных.
Адсорбционная теория пассивности – пассивность вызывается адсорбционными плёнками кислорода. Она не может объяснить явление перепассивации, но легко объясняет влияние СL-, он является поверхностно активным ионом, поэтому он сам адсорбируется и вытесняет кислород. Плёнка СL- не тормозит коррозионный процесс. На некоторых Ме оксидов не обнаруживается при ЕПАССИВ.
Сейчас пассивность объясняется теорией смешанной пассивности: за начало пассивации ответственна адсорбция, потом эти слои преобразуются в оксидные и гидроксидные слои. Оксид и гидроксид тоже способствуют пассивации. Даже если оксид и гидроксид образуются на всей поверхности Ме – адсорбция продолжает играть роль в пассивации.
![]() Оксид
и гидроксид имеют поры, в них адсорбируется
кислород и за счёт одновременного
действия плёнки поверхность Ме пассивна.
При попадании в коррозионную среду
СL-, он не может разрушить оксид, но может
вытеснить адсорбированный кислород,
начинается питтинговая коррозия (AL,
сталь – пассивны в
HNO3).
Оксид
и гидроксид имеют поры, в них адсорбируется
кислород и за счёт одновременного
действия плёнки поверхность Ме пассивна.
При попадании в коррозионную среду
СL-, он не может разрушить оксид, но может
вытеснить адсорбированный кислород,
начинается питтинговая коррозия (AL,
сталь – пассивны в
HNO3).
Солевая пассивация возникает, если в коррозионной среде присутствуют анионы, способные давать труднорастворимую соль с корродирующим Ме(пассивация в Н2SO4 FeSO4, при высоких концентрациях Н2SО4 растворимость FeSO4 снижается – образуется солевая упорядоченная плёнка). Солевой пассивацией объясняется устойчивость Fe в фосфатах, устойчивость свинца в Н2SO4.
Использование пассивности в практике защиты:
Вводят в коррозионную среду окислители, столько, чтобы защищаемый Ме был полностью запассивирован за счёт торможения анодного процесса.
Понижают анодную активность Ме путём легирования его более легко пассивирующимися Ме (железо хромом>13%).
Применяют катодные протекторы – контактирование защищаемого Ме с более эл.положительными Ме и т.д.
Анодные процессы электрохимической коррозии. Продукты коррозии. Диаграммы Пурбе.
Анодные процессы электрохимической коррозии для двух валентного Ме идут по 7 направлениям:
1)Ме гидратируются с образованием гидратированных катионов: Ме-2ё=>Ме2+
2)Адсорбционные плёнки О2: Ме-2ё+Н2О=>МеО+2Н+
3)Ме ионизируются с образованием оксидов: Ме-2ё+Н2О=>МеО+2Н+
4)С образованием гидроксидов: Ме-2ё+2Н2О=>Ме(ОН)2+2Н+
5)С образованием сложных анионов: Ме-3ё-2Н2О=>МеО2-+4Н+
6)С образованием комплексных ионов: Ме-2ё =>[Ме(NH3)n]2+ под действием катиона NH3
Ме-2ё =>[Ме(CN)n](n-2) под действием аниона CN-
7)С образованием труднорастворимых солей: Ме-2ё+2А-=>МеА2
В настоящее время точный механизм анодных реакций коррозионного процесса неизвестен для большинства Ме (для Fe существует 6 механизмов):
А) Каждая из этих реакций идёт по стадиям и в отдельных стадиях могут участвовать анионы коррозионной среды: Fe H2SO4, HCL
Fe – 2ё=>Fe2+
Б) Анодная реакция может протекать по двум-трём направлениям, поэтому получаются сложные продукты коррозии: CuCL2, 3Cu(OH)3. Анодная реакция может протекать с образованием растворимых продуктов коррозии по направлению 1, 5, 6, тормозящего действия нет.
Нерастворимые продукты коррозии образуются по направлениям 2, 3, 4, 7 и скорость эл.химической коррозии зависит от того, какие это продукты – упорядоченные или нет, плотные или неплотные.
Чтобы узнать по какому пути идёт реакция необходимо: узнать состав коррозионной среды, рН и потенциал коррозии. Если в коррозионной среде присутствуют комплексообразователи, способные давать комплекс с корродирующим Ме, то реакция идёт по направлению 6, если нет комплексообразователя, но есть анионы, способные давать с корродирующим Ме труднорастворимую соль, то реакция идёт по направлению 7, если нет ни того ни другого, то обращаются к диаграммам Пурбе для данного Ме, они связывают наиболее термодинамически устойчивое состояние Ме (оксид, гидроксид) с его потенциалом коррозионной среды.
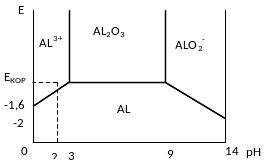
Положение линий зависит от концентрации АL+. При известном потенциале коррозии и рН определяем направление реакции в зависимости сколько воды в 7-ми реакциях. В нижней части системы АL – H2O, алюминий устойчив и не подвержен коррозии. В области АL3+ он не устойчив и будет корродировать с образованием АL3+(также корродирует в других областях). По этой диаграмме АL - амфотерный Ме.
Атмосферная коррозия металлов, основные особенности. Оценка коррозионной агрессивности атмосферы.
Под действием окружающего нас воздуха. Под ней понимают любую коррозию. Самый распространенный вид коррозии (80%). Скорость зависит от толщины пленки влаги на поверхности.
1)Сухая атмосферная коррозия (хим.) 2)Влажная атмосферная коррозия. 3)Мокрая атмосферная коррозия. 4)Подводная коррозия.
Учитывая опасность
коррозии, выпустили ГОСТ 9.039-74 «Коррозионная
агрессивность атмосферы». Оценивается
несколькими параметрами: 1. Время
увлажнения адсорбционной пленкой влаги
( ).
2. Время увлажнения фазовой пленкой
(
).
2. Время увлажнения фазовой пленкой
( ).
3. Общее время увлажнения
).
3. Общее время увлажнения
 .
4. Концентрация коррозионно-агессивных
компонентов
.
4. Концентрация коррозионно-агессивных
компонентов
 .
.
По величине
 ГОСТ все атмосферы делит на 9 баллов
коррозионной агрессивности:
ГОСТ все атмосферы делит на 9 баллов
коррозионной агрессивности:
|
Балл |
500-1000 ч/год |
1 |
1000- 1500 ч/год |
2 |
>4500 ч/год |
9 |
Потери от атмосферной коррозии могут быть оценены с помощью ГОСТ- 9.040- 74 «Расчетно- экспериментальный метод оценки потерь от атмосферной коррозии». Согласно ему потери Ме в первый год:
 ( г/м2) , где
К
( г/м2) , где
К -
-
 коррозии
под адсорбционной пленкой влаги;
коррозии
под адсорбционной пленкой влаги;
 -
коэф. учитывающий увеличение
-
коэф. учитывающий увеличение
 под
адсорбционной пленкой влаги за счет
активного компонента с концентрацией
С;
под
адсорбционной пленкой влаги за счет
активного компонента с концентрацией
С;
 -
под
фазовой пленкой влаги;
-
под
фазовой пленкой влаги;
 -
коэф. учитывающий увеличение
под фазовой пленкой влаги за счет
коррозионно- активного компонента с
концентрацией С. Они определяются по
методике предложенной в ГОСТе для
разных атмосфер.
-
коэф. учитывающий увеличение
под фазовой пленкой влаги за счет
коррозионно- активного компонента с
концентрацией С. Они определяются по
методике предложенной в ГОСТе для
разных атмосфер.
За несколько лет:
 ,
где n- коэффициент, учитывающий влияние
продуктов коррозии. В зависимости от
вида компонентов которые присутствуют
в атмосфере различают несколько видов:
1. Сельская 2. Городская 3. Промышленная
4. Тропическая 5. Морская. Самыми активными
считаются промышленная и тропическая
атмосферы.
,
где n- коэффициент, учитывающий влияние
продуктов коррозии. В зависимости от
вида компонентов которые присутствуют
в атмосфере различают несколько видов:
1. Сельская 2. Городская 3. Промышленная
4. Тропическая 5. Морская. Самыми активными
считаются промышленная и тропическая
атмосферы.
Методы борьбы с коррозией: 1. Сушка 2. Силикагель 3. Смазка, покрытия. 4. Легирование.
Поведение железа и его сплавов в условиях химической и электрохимической коррозии.
Химической корроз называется процесс самопроизв разруш Ме под действием сухих газов и неэлектролитов, при котором окисл Ме и восстановл окислит протекают в виде одного элементарного акта. Наиб распростр газовая корроз под действ воздуха (основн окисл кислород, дополнит- серовод, диоксид серы). Железо корродир с кислород и протек гетерог хим реакц в результ которой на поверхн появл оксидн пленка (другие окисл приводят к появл солевых пленок).
Fe дает три вида оксидов: Fe2О3- гематит; Fe3О4-магнетит; FeО-вюстит.
При низких температурах до 4000С обрзуется -Fe2О3 пленка упорядоченная наблюдается логарифмический закон. От 400-5750С -Fe3О4 наблюдается степенной закон, выше 5750С-FeО-вюстит(параболический закон). 575 температура окалинообразования Fe в чистом воздухе.
схема окалины:
2. Если в воздухе появляется 5% воды то FeО начинает образовываться при 4270С.
3. Если в воздухе появляется 5% воды и 5% SО2, то FeО начинает образовываться при 3000С.
В реальной практике мы имеем 3 стадию, поэтому 3000С – температура окалинообразования железа.
Таким образом железо и его сплавы можно не защищать в условиях хим. коррозии.
Электрохимической коррозией называется называется самопроизвольный процесс разрушения Ме под действием электролитов, при котором окисление Ме и восстановление окислителя протекает в виде двух сопряженных электрохимич реакций. Процесс электрохим корроз наблюд всегда, когда на поверхности Ме появл электролит.
Fе не очень склонно к комплексообразованию; Fе часто дает трудно растворимые соли FеСО3, если появляются SiО3-, Р2 О5-, то коррозия идет по пути увеличения. Если нет ни того ни другого обращаемся к диаграмме Пурбе:
Кисл. среды В кисл растворах с рН<3 сплавы железа корродируют с образов ионов Fe2+,т.е. получ растворимые продукты коррозии.
Окислителями выступ как Н+, так и растворимый в корроз среде О2, в окисляющих кислотах окислителями выступают анионы этих кисл. в слаб раствор кислот увелич скор корроз (рис 2) связан не только с рост концентр Н+, но и с улучш диффузии О2 к поверх Ме,т.к. естест диффуз-барьерн оксид пленка в раств таких кисл полностью сним. Прич в сильн кислотах пленка расв при рН=3-4, а в более слаб при рН=5-6.
Щелоч среды. В щелоч раств с 11,5<рН<13 согл диагр Пурбе, также получ гидроксиды железа, но они получ упорядоч и хорошо защищ металл. Принято считать, что сталь и чугун стойки в разбавл раств щелочей. Однако в прис Cl- может набл локал корроз (питтинговая). В конц раств щелоч анодн. реакц корроз процесса идет с образ феррит-ионов HFeО2- и корроз вновь станов блее опасн. Кипящ растворы едкого натра опасны при конц свыше10% .
Нейтрал среды. В раствор с 3<рН<11,5 в качеств продукт корроз получ в начале гидрокс Fе(ОН)2, далее он окисл до Fе(ОН)3. Fе-2е+2 ОН-→ Fе(ОН)2→ Fе(ОН)3 Смесь двух гидроксид это ржавчина. В дальнейш при недостатке влоги происх дегидротац продукт корроз с образован обычн бурой ржавч
2Fе(ОН)3+2 Н2О→ Fе2 О3• Н2О
При налич в воде и атмосф СО2, Н2S, SО2 в составе ржавч появл соедин FеСО3, FеSО4, FеS и эти компон упорядочив образующ. пленку или наоборт делают ее менее плотной.Скор корроз в нейтр средах, где она контрол диффуз кислор, удваив при повыш темпер на кажд 300С.Повыш идет вплоть до 800С, когда скор корроз начинает падать из-за уменьш раствор кислорода.Таким образом железо в условиях эл.химич коррозии необходимо защищать во всех средах кроме щелочных.


