

1
Химия и материя. Частицы материи: элементарные частицы, атомы, молекулы, продукты их ассоциации и агрегации. Вопросы химической метрологии. Современная система атомных масс. Относительные атомные и молекулярные массы. Количество вещества. Молярная масса. Молярный обьем.
ХИМИЯ, наука, изучающая строение в-в и их превращения, сопровождающиеся изменением состава и(или) строения. Хим. св-ва в-в (их превращения) определяются гл. обр. состоянием внеш. электронных оболочек атомов и молекул, образующих в-ва; состояния ядер и внутр. электронов в хим. процессах почти не изменяются. Объектом хим. исследований являются химические элементы и их комбинации, т. е. атомы, простые (одноэлементные) и сложные (молекулы, ионы, ион-радикалы, свободные радикалы) хим. соед., их объединения (ассоциаты, сольваты и т. п.), материалы и др. Число хим. соед. огромно и все время увеличивается; поскольку химия сама создает свой объект исследования.
Материя в химии представляется в виде атомов, атомных ядер и электронов, обладающих свойством образования между собой химических связей при соединении в молекулы.
Элементарные частицы - частицы, не имеющие, как правило, внутренней структуры (т.е. не содержащая других частиц). Обычно к ним относят составные части атома: электроны и ядро атома
Для химии наибольшее значение имеют такие элементарные частицы, как электроны, протоны и нейтроны, из которых образованы атомы химических элементов.
Электрон (первый, из открытых элементарных частиц) — стабильная элементарная частица с отрицательным зарядом, равным элементарному электрическому заряду. (кванту электричества) Число электронов в атоме определяет его поведение в химических реакциях
Ядро атома - состоит из протонов и нейтронов. Пространство между ядром и электронами заполнено электромагнитным полем. Если число протонов в ядре равно числу электронов, атом в целом электрически нейтрален. В противном случае он обладает некоторым избыточным (положительным или отрицательным) зарядом и называется ионом. Количество протонов в ядре определяет принадлежность атома некоторому химическому элементу, а количество нейтронов - изотопу этого элемента
Нейтрон - элементарная частица, входящая в состав ядра атома, не имеющая электрического заряда (нейтральна). Масса нейтрона намного больше массы электрона
Протон - элементарная частица, являющаяся составной частью ядер атомов всех химических элементов. Заряд протона положителен (1.6▪10-19Кулон); он равен заряду электрона, но противоположен ему по знаку
Атом (в переводе с греческого означает неделимый) —система элементарных частиц, состоящая из- ядра, образованного протонами и нейтронами, и электронов. (одноядерная система, с отличной от нуля массой. )Это наименьшая частица химического элемента, носитель его свойств и способная к самостоятельному существованию. При любом многократном механическом дроблении вещества (например, кусочка золота) даже самые мельчайшие из них все равно обладают всеми свойствами этого металла. Атомы могут образовывать более крупные частицы - молекулы, состоящие из двух или большего количества атомов Каждому химическому элементу соответствует совокупность определенных атомов.
Все химические частицы, образуемые атомами элементов и участвующие в химических процессах, могут быть классифицированы на:
■ простые: ♦ электроны ♦ ядра атомов ♦ атомы и их изотопы
■ сложные: ♦ молекулы (в т.ч. изомеры):
▪ простых веществ
▪ сложных веществ: ▫ низкомолекулярных ▫ высокомолекулярных
♦ радикалы ♦ ионы: ▪ атомные:
▫ анионы
▫ катионы
▪ молекулярны ▪ комплексные
♦ комплексные частицы
Молекула (новолат. molecula, уменьшит. от лат. moles – масса), микрочастица, образованная из двух или большего числа атомов и способная к самостоятельному существованию. Имеет постоянный состав (качественный и количественный) входящих в нее атомных ядер и фиксированное число электронов и обладает совокупностью свойств, позволяющих отличать одну молекулу от других, в т.ч. от молекул того же состава. Молекулы состоят из двух или большего количества атомов одного или различных химических элементов, объединенных межатомными связями (иначе: молекула - это устойчивая группа атомов, связанных химическими связями). Для одноатомных молекул (например, благородных газов) понятие 'атом' и 'молекула' совпадают
Ионы (от греч. ion – идущий), одноатомные или многоатомные частицы, несущие электрический заряд. Положительные ионы называют катионами (от греч. kation, буквально – идущий вниз), отрицательные – анионами (от греч. anion, буквально
идущий вверх). В свободном состоянии существуют в газовой фазе (в плазме).
Метроло́гия (от греч μέτρον — мера, измерительный инструмент + др.-греч. λόγος — мысль, причина) — наука об измерениях, методах и средствах обеспечения их единства и способах достижения требуемой точности Предметом метрологии является извлечение количественной информации о свойствах объектов с заданной точностью и достоверностью; нормативная база для этого — метрологические стандарты
Метрология состоит из 3 разделов:
Теоретическая - Рассматривает общие теоретические проблемы (разработка теории и проблем измерений физических величин, их единиц, методов измерений).
Прикладная - Изучает вопросы практического применения разработок теоретической метрологии. В её ведении находятся все вопросы метрологического обеспечения.
Законодательная - Устанавливает обязательные технические и юридические требования по применению единиц физической величины, методов и средств измерений.
В 1961 г. принята единая шкала относительных атомных масс, в основу которой положена 1/12 часть массы атома изотопа углерода 12C, названная атомной единицей массы (а.е.м.). В соответствии с этим в настоящее время относительной атомной массой (сокращенно — атомной массой) элемента называют отношение массы его атома к 1/12 части массы атома 12C . В современной шкале относительные атомные массы кислорода и водорода равны соответственно 15,9994 и 1,00794.
Аналогично относительной молекулярной массой (сокращенно — молекулярной массой) простого или сложного вещества называют отношение массы его молекулы к 1/12 части массы 12C . Поскольку масса любой молекулы равна сумме масс составляющих ее атомов, то относительная молекулярная масса равна сумме соответствующих относительных атомных масс. До недавнего времени вместо терминов «атомная масса» и «молекулярная масса» употреблялись термины «атомный вес» и «молекулярный вес».
Наряду
с единицами массы и объема в химии
пользуются также единицей количества
вещества, называемой молем (сокращенно
обозначение — «моль»). Моль
— количество вещества,
содержащее столько молекул, атомов,
ионов, электронов или других структурных
единиц, сколько содержится атомов в 12
граммах изотопа
углерода 12С.
Применяя понятие «моль», необходимо в
каждом конкретном случае точно указывать,
какие именно структурные единицы имеются
в виду. Например, следует различать моль
атомов Н, моль молекул Н2,
моль ионов Н+.
В настоящее время число структурных
единиц, содержащихся в одном моле
вещества (постоянная Авогадро), определено
с большой точностью. В практических
расчетах его принимают равным 6,02*1023.
Отношение массы m вещества к его
количеству n
называют
молярной массой вещества
![]() Молярную массу обычно выражают в г/моль.
Поскольку в одном моле любого вещества
содержится одинаковое число структурных
единиц, то молярная масса вещества M
, г/моль) пропорциональна массе
соответствующей структурной единицы,
т. е. относительной молекулярной (или
атомной) массе данного вещества (Мотн)
Молярную массу обычно выражают в г/моль.
Поскольку в одном моле любого вещества
содержится одинаковое число структурных
единиц, то молярная масса вещества M
, г/моль) пропорциональна массе
соответствующей структурной единицы,
т. е. относительной молекулярной (или
атомной) массе данного вещества (Мотн)
![]() где К — коэффициент пропорциональности,
одинаковый для всех веществ. Mолярная
масса вещества, выраженная в граммах
на моль, имеет то же численное значение,
что и его относительная молекулярная
(атомная) масса. Согласно закону Авогадро
( в равных обьемах любых газов, взятых
при одной и той же температуре и при
одинаковом давлении, содержится одно
и тоже число молекул) одно и то же число
молекул любого газа занимает при
одинаковых условиях один и тот же объем.
С другой стороны, 1 моль любого вещества
содержит (по определению) одинаковое
число частиц. Отсюда следует, что при
определенных температуре и давлении 1
моль любого вещества в газообразном
состоянии занимает один и тот же объем.
Нетрудно рассчитать, какой объем занимает
один моль газа при нормальных условиях,
т. е. при нормальном атмосферном
давлении 760 мм рт ст ) и
температуре 00С. Например, экспериментально
установлено, что масса 1 л кислорода при
нормальных условиях равна 1,43 грамм.
Следовательно, объем, занимаемый при
тех же условиях одним молем кислорода
(32 грамм), составит 32:1,43=22,4 л. То же число
получим, рассчитав объем одного моля
водорода, диоксида углерода и т. д.
Отношение объема, занимаемого веществом,
к его количеству называется молярным
объемом вещества. Как следует из
изложенного, при нормальных условиях
молярный объем (Vm )любого газа равен
22,4 л/моль. Vm = V /n
где К — коэффициент пропорциональности,
одинаковый для всех веществ. Mолярная
масса вещества, выраженная в граммах
на моль, имеет то же численное значение,
что и его относительная молекулярная
(атомная) масса. Согласно закону Авогадро
( в равных обьемах любых газов, взятых
при одной и той же температуре и при
одинаковом давлении, содержится одно
и тоже число молекул) одно и то же число
молекул любого газа занимает при
одинаковых условиях один и тот же объем.
С другой стороны, 1 моль любого вещества
содержит (по определению) одинаковое
число частиц. Отсюда следует, что при
определенных температуре и давлении 1
моль любого вещества в газообразном
состоянии занимает один и тот же объем.
Нетрудно рассчитать, какой объем занимает
один моль газа при нормальных условиях,
т. е. при нормальном атмосферном
давлении 760 мм рт ст ) и
температуре 00С. Например, экспериментально
установлено, что масса 1 л кислорода при
нормальных условиях равна 1,43 грамм.
Следовательно, объем, занимаемый при
тех же условиях одним молем кислорода
(32 грамм), составит 32:1,43=22,4 л. То же число
получим, рассчитав объем одного моля
водорода, диоксида углерода и т. д.
Отношение объема, занимаемого веществом,
к его количеству называется молярным
объемом вещества. Как следует из
изложенного, при нормальных условиях
молярный объем (Vm )любого газа равен
22,4 л/моль. Vm = V /n
2
Основные законы химии :закон сохранения массы вещества и энергии, закон постоянства состава, закон кратных отношений; закон простых объемных отношений; закон Авогадро и его следствия. Методы определение атомных масс элементов. Методы определения молекулярных масс
закон сохранения массы вещества и энергии
После доказательства существования атомов и молекул важнейшим открытием атомно-молекулярной теории стал закон сохранения массы, который был сформулирован в виде философской концепции русским ученым Михаилом Васильевичем Ломоносовым в 1748 г. и подтвержден экспериментально им самим в 1756 г. и независимо от него французским химиком А.Л.Лавуазье в 1789 г.
Масса всех веществ, вступающих в химическую реакцию, равна массе всех продуктов реакции.
В химической реакции число взаимодействующих атомов остается неизменным, происходит только их перегруппировка с разрушением исходных веществ. Взаимодействие водорода и кислорода с образованием воды может быть записано с помощью уравнения химической реакции
|
|
|
Коэффициенты перед формулами химических соединений называются стехиометрическими.
Закон сохранения массы является частным случаем общего закона природы - закона сохранения энергии, который утверждает, что энергия изолированной системы постоянна. Энергия - это мера движения и взаимодействия различных видов материи. При любых процессах в изолированной системе энергия не производится и не уничтожается, она может только переходить из одной формы в другую.Одной из форм энергии является так называемая энергия покоя, которая связана с массой соотношением Эйнштейна Е0 = m0•с2, где с - скорость света в вакууме (с = 3•108 м/с). Это соотношение показывает, что масса может переходить в энергию и наоборот. Именно это и происходит во всех ядерных реакциях, и поэтому закон сохранения массы в ядерных процессах нарушается. Однако, закон сохранения энергии остается справедливым и в этом случае, если учитывать энергию покоя. В химических реакциях изменение массы, вызванное выделением или поглощением энергия, очень мало. закон постоянства состава Всякое чистое вещество независимо от способа его получения всегда имеет постоянный качественный и количественный состав.
Атомно-молекулярное учение позволяет объяснить закон постоянства состава. Поскольку атомы имеют постоянную массу, то и массовый состав вещества в целом постоянен.
Закон постоянства состава впервые сформулировал французский ученый-химик Ж.Пруст в 1808 г. Он писал: "От одного полюса Земли до другого соединения имеют одинаковый состав и одинаковые свойства. Никакой разницы нет между оксидом железа из Южного полушария и Северного. Малахит из Сибири имеет тот же состав, как и малахит из Испании.".
В этой формулировке закона, как и в приведенной выше, подчеркивается постоянство состава соединения независимо от способа получения и места нахождения.
Развитие химии показало, что наряду с соединениями постоянного состава существуют соединения переменного состава. По предложению Н.С. Курнакова первые названы дальтонидами (в память английского химика и физика Дальтона), вторые -бертоллидами (в память французского химика Бертолле, предвидевшего такие соединения). Состав дальтонидов выражается простыми формулами с целочисленными стехиометрическими индексами, например Н2О, НCl, ССl4, СO2. Состав бертоллидов изменяется и не отвечает стехиометрическим отношениям.
В связи с наличием соединений переменного состава в современную формулировку закона постоянства состава возможно внести уточнение.
Cостав соединений молекулярной структуры, т.е. состоящих из молекул, - является постоянным независимо от способа получения. Состав же соединений с немолекулярной структурой (с атомной, ионной и металлической решеткой) не является постоянным и зависит от условий получения. Однако условно для простоты состав многих бертоллидов записывают как постоянный. Например, состав оксида железа(II) записывают в виде FeO (вместо более точной формулы Fe1-xO).
закон кратных отношений
Если два элемента образуют между собой несколько молекулярных соединений, то масса одного элемента, приходящаяся на одну и ту же массу другого, относятся между собой как небольшие целые числа.
При взаимодействии азота с кислородом образуются пять оксидов. На 1 грамм азота в образующихся молекулах приходится 0,57, 1,14, 1,71, 2,28, 2,85 грамм кислорода, что соответствует отношением 2:1, 1:1, 2:3, 1:2, 2:5 в этих оксидах; их составы N2O, NO, N2O3, NO2, N2O5.
Закон кратных отношений открыт в 1803 Дж.Дальтоном и истолкован им с позиций атомизма.
закон простых объемных отношений
(открыт Ж. Гей-Люссаком):
При равных условиях объемы вступающих в реакцию газов относятся друг к другу и к объемам образующихся газообразных продуктов, как небольшие целые числа.
Так, в реакции образования аммиака из простых веществ отношение объемов водорода, азота и аммиака составляет 3 : 1 : 2.
Закон Авогадро: В равных объемах любых газов, взятых при одинаковых условиях, содержится одинаковое число молекул.
Из закона Авогадро вытекают два следствия:
Одинаковое количество молекул любых газов при одинаковых условиях занимают одинаковый объем. В частности, при нормальных условиях, т. е. при 0 °C (273К) и 101,3 кПа, объём 1 моля газа, равен 22,4 л. Этот объём называют молярным объёмом газа Vm. Пересчитать эту величину на другие температуру и давление можно с помощью уравнения Менделеева-Клапейрона:
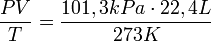 .
.Относительная плотность одного газа по другому равна отношению их молярных масс.
Положение это имело громадное значение для развития химии, так как оно дает возможность определять частичный вес[2]тел, способных переходить в газообразное или парообразное состояние. Если через m мы обозначим частичный вес тела, и через d — удельный вес[3] его в парообразном состоянии, то отношение m / d должно быть постоянным для всех тел. Опыт показал, что для всех изученных тел, переходящих в пар без разложения, эта постоянная равна 28,9, если при определении частичного веса исходить из удельного веса воздуха, принимаемого за единицу, но эта постоянная будет равняться 2, если принять за единицу удельный вес водорода. Обозначив эту постоянную, или, что то же, общий всем парам и газам частичный объём через С, мы из формулы имеем с другой стороны m = dC. Так как удельный вес пара определяется легко, то, подставляя значение d в формулу, выводится и неизвестный частичный вес данного тела.
Число Авогадро – число частиц в моле любого вещества; NA = 6,02∙1023 моль–1.
Молярный объем – объем моля любого газа при нормальных условиях(температура 273 К, давление 101,3 кПа); равен 22,4 л∙моль–1.
Молярная масса (M) – масса одного моля вещества, численно совпадающая с относительными массами атомов, ионов, молекул, радикалов и других частиц, выраженных в г∙моль–1.
Некоторые методы определения атомных масс химических элементов
1. Метод Авогадро
Как было показано закон Авогадро позволяет определить молекулярные массы газов. С другой стороны результаты изучения объемных соотношений газов, вступающих в реакцию, и полученных газообразных продуктов позволяют определить число атомов в молекуле.
Так, при взаимодействии 1 объема хлора с 41 объемом водорода образуются 2 объема хлороводорода. Из этого следует, что молекула хлора, как и молекула водорода, состоит из 2-х атомов.
Аналогично было установлено, что молекулы других простых газов, таких, как кислород, азот также двухтомны. Атомную массу перечисленных газов находят делением их молекулярной массы на 2.
Например, молекулярная масса хлора равна 71, следовательно, его атомная масса 35,5.
О количестве атомов, входящих в состав молекул, можно также судить по их молярной теплоемкости. Именно по результатам измерения этой характеристики благородных газов было установлено, что их молекулы одноатомны и атомная масса этих газов равна их молекулярной массе.
2. Метод Канниццаро
Этот метод применим для определения атомных масс элементов, дающих газообразные или легколетучие соединения.
Для нахождения атомной массы этим методом определяют молярную массу возможно большего числа газообразных или легколетучих соединений данного элемента. Затем на основании данных анализа рассчитывают, сколько атомных единиц массы приходится на долю этого элемента в молекуле каждого из взятых соединений. Наименьшее количество данного элемента в молекуле изученных веществ и будет его атомной массой, так как в молекуле не может находиться меньше 1 атома.
В табл. 2 приведены молекулярные массы ряда соединений углерода, процентное содержание углерода в каждом из них, а также масса углерода, содержащаяся в каждом из этих соединений.
Наименьшая масса углерода, содержащегося в молекулах приведенных соединений равна 12 а.е.м. Следовательно, атомная масса углерода не может быть больше 12 (иначе пришлось бы предположить, что в состав сероуглерода, диоксида и монооксида углерода входит часть атома углерода). Считать атомную массу углерода меньше 12 нет оснований, так как соединения, содержащие менее 12 а.е.м. углерода, неизвестны.
3. Метод Менделеева
Атомную массу элемента можно рассчитать, исходя из положения этого элемента в Периодической системе. Приблизительную атомную массу элемента можно вычислить как среднеарифметическое атомных масс соседних с ним элементов. Так атомная масса алюминия, рассчитанная как среднеарифметическое атомных масс магния, кремния, бора и скандия равна
Ar(Mg)+Ar(Si)+ Ar(B)+ Ar(Sc))/4=(24,3+28,08+10,8+44,95)/4=27,03 что вполне удовлетворительно согласуется с табличной величиной 26,98. Для определения точного значения атомной массы элемента необходимо знать его эквивалентную массу. Разделив приблизительное значение атомной массы элемента на его эквивалентную массу, находят валентность элемента, округлив ее до целочисленного значения, и затем, умножая эквивалентную массу на валентность элемента, находят его точную атомную массу.
4. Метод Дюлонга и Пти
Французские ученые П. Дюлонг и А. Пти установили закон, согласно которому атомная теплоемкость простого вещества в твердом состоянии (т. е. произведение его удельной теплоемкости на молярную массу атомов) есть величина постоянная и равная в среднем 26 Дж/ (К(моль).
Из закона Дюлонга и Пти следует, что, разделив 26 на удельную теплоемкость простого вещества, легко определяемую экспериментально, можно найти приблизительное значение атомной массы данного элемента.
Чтобы перейти от приблизительного значения атомной массы к точному ее значению, предварительно определяют опытном путем эквивалентную массу данного элемента. Разделив приблизительное значение атомной массы на его эквивалентную массу, находят валентность элемента, часто несколько отличающуюся от целого числа.
Так как валентность выражается только целыми числами, найденное значение округляют. Умножив эквивалентную массу на валентность, получают точное значение мольной массы атомов.
Методы определения молекулярных масс
Исторически первый метод (обоснованный исследованиями С. Канниццаро и А. Авогадро) предложен Ж. Дюма в 1827 и заключался в измерении плотности газообразных веществ относительно водородного газа, молярная масса к-рого принималась первоначально равной 2, а после перехода к кислородной единице измерений молекулярных и атомных масс-2,016 г. След. этап развития эксперим. возможностей определения молекулярной массы заключался в исследовании жидкостей и р-ров нелетучих и недиссоциирующих в-в путем измерения коллигативных св-в (т. е. зависящих только от числа растворенных частиц) - осмотич. давления (Осмометрия), понижения давления пара, понижения точки замерзания (криоскопия)и повышения точки кипения (эбулиоскопия)р-ров по сравнению с чистым р-рителем. При этом было открыто "аномальное" поведение электролитов.
Понижение давления пара над
р-ром зависит от молярной доли растворенного
в-ва (закон Рауля): [(р - р0)/р] = N, где
р0-давление пара чистого р-рителя,
р-давление пара над р-ром, N- молярная
доля исследуемого растворенного в-ва,
N = (тх/Мх)/[(тх/Мх) + (m0/M0)], mx и Мх-соотв.
навеска (г) и молекулярная масса
исследуемого в-ва, m0 и М0-то же для
р-рителя. В ходе определений проводят
экстраполяцию к бесконечно разб. р-ру,
т.е. устанавливают ![]() для
р-ров исследуемого в-ва и для р-ров
известного (стандартного) хим.
соединения. В случае криоскопии и эбулиоскопии используют
зависимости соотв. Dtз = Кс и Dtк =
Еc, где Dtз-понижение т-ры замерзания
р-ра, Dtк - повышение т-ры кипения р-ра,
К и Е-соотв. криоскопич. и эбулиоскопич.
постоянные р-рителя, определяемые по
стандартному растворенному в-ву с точно
известной молекулярной массой,
с-моляльная концентрация исследуемого
в-ва в р-ре (с = Мхтх.1000/m0). Молекулярную
массу рассчитывают по ф-лам: Мвва =
тхК.1000/m0Dt3 или Мх = тхЕ.1000/m0 Dtк.
Методы характеризуются достаточно
высокой точностью, т.к. существуют
спец. термометры (т. наз.термометры
Бекмана), позволяющие измерять весьма
малые изменения т-ры.
для
р-ров исследуемого в-ва и для р-ров
известного (стандартного) хим.
соединения. В случае криоскопии и эбулиоскопии используют
зависимости соотв. Dtз = Кс и Dtк =
Еc, где Dtз-понижение т-ры замерзания
р-ра, Dtк - повышение т-ры кипения р-ра,
К и Е-соотв. криоскопич. и эбулиоскопич.
постоянные р-рителя, определяемые по
стандартному растворенному в-ву с точно
известной молекулярной массой,
с-моляльная концентрация исследуемого
в-ва в р-ре (с = Мхтх.1000/m0). Молекулярную
массу рассчитывают по ф-лам: Мвва =
тхК.1000/m0Dt3 или Мх = тхЕ.1000/m0 Dtк.
Методы характеризуются достаточно
высокой точностью, т.к. существуют
спец. термометры (т. наз.термометры
Бекмана), позволяющие измерять весьма
малые изменения т-ры.
Для определения молекулярной массы используют также изотермич. перегонку р-рителя. При этом пробу р-ра исследуемого в-ва вносят в камеру с насыщ. паром р-рителя (при данной т-ре); пары р-рителя конденсируются, т-ра р-ра повышается и после установления равновесия вновь понижается; по изменению т-ры судят о кол-ве выделившейся теплоты испарения, к-рая связана с молекулярной массой растворенного в-ва. В т. наз. изопиестич. методах проводят изотермич. перегонку р-рителя в замкнутом объеме, напр. в Н-образном сосуде. В одном колене сосуда находится т. наз. р-р сравнения, содержащий известную массу в-ва известной молекулярной массы (молярная концентрация C1), в другом-р-р, содержащий известную массу исследуемого в-ва (молярная концентрация С2 неизвестна). Если, напр., С1 > С2, р-ритель перегоняется из второго колена в первое, пока молярные концентрации в обоих коленах не будут равны. Сопоставляя объемы полученных изопиестич. р-ров, рассчитывают молекулярную массу неизвестного в-ва. Для определения молекулярной массыы можно измерять массу изопиестич. р-ров с помощью весов Мак-Бена, к-рые представляют собой две чашечки, подвешенные на пружинках в закрытом стеклянном сосуде; в одну чашечку помещают исследуемый р-р, в другую-р-р сравнения; по изменению положения чашечек определяют массы изопиестич. р-ров и, следовательно, молекулярную массу исследуемого в-ва.
Осн. методом определения атомных и мол. масс летучих в-в является масс-спектрометрия. Для исследования смеси соед. эффективно использование хромато-масс-спектромет-рии. При малой интенсивности пика мол.иона применяют эффузиометрич. приставки к масс-спектрометрам. Эффузио-метрич. способ основан на том, что скорость вытекания газа в вакуум из камеры через отверстие, диаметр к-рого значительно меньше среднего пути своб. пробега молекулы, обратно пропорциональна квадратному корню из молекулярной массы в-ва; скорость вытекания контролируют по изменению давления в камере. Молекулярную массу летучих соед. определяют также методами газовой хроматографии с газовыми весами Мартина. Последние измеряют скорость перемещения газа в канале, соединяющем трубки, по к-рым текут газ-носитель и газ из хроматографич. колонки, что позволяет определять разницу плотностей этих газов, зависящую от молекулярной массы исследуемого в-ва.
Средние значения молекулярных масс полимеров устанавливают с помощью перечисленных выше методов, основанных на коллигативных св-вах разбавленных р-ров, по числу двойных связей ("мягким" озонолизом) или функц. групп (методами функцион. анализа), а также по таким св-вам их р-ров, как вязкость,светорассеяние Средние значения мол. масс полимеров высокой степени полимеризации определяют по их реологич. характеристикам.
3
Основные положения квантовой механики. Волновые и корпускулярные свойства микрочастиц. Гипотеза де Бройля. Волновая функция. Соотношение неопределенностей Гейзенберга. Квантовые числа Их физический смысл при характеристике электронного строения атомов.
В основе идеи квантовой механики лежит представление о двойственности природы света. Если в классической теории электрону приписывают только корпускулярные свойства, а свет рассматривают как электромагнитную волну, то в квантовой механике электрон обладает как корпускулярными, так и волновыми свойствами.
Разногласия классической и квантовой теорий:
1) Фотоэффект, эффект Комптона. Фотоэффект бывает внутренний и внешний:
внешний фотоэффект – вырывание электронов с поверхности твердых и жидких тел в результате действия света;
2) Устойчивость атомов. С позиции классической физики электрон, вращающийся вокруг атомного ядра, должен непрерывно испускать энергию, терять скорость и падать на ядро. Т.е. стационарное состояние планетарной модели атома невозможно (частота обращения должна постоянно меняться), а совокупность атомов должна давать непрерывный спектр излучения.
В 1913 году Нильс Бор в теории о строении атома сформулировал два постулата:
1) Постулат стационарных орбит (состояний) – существуют определенные стационарные состояния атома, находясь в которых он не излучает энергию.
2) Постулат частот – при переходе из одного стационарного состояния в другое атом излучает/поглощает квант электромагнитной энергии – фотон.
Как у света, так и у микрочастиц мы не можем одновременно наблюдать проявление волновых и корпускулярных свойств.
В 1924 г. Луи де Бройль выдвинул гипотезу об универсальности корпускулярно-волнового дуализма. Согласно этой гипотезе не только фотоны, но и любые другие частицы материи наряду с корпускулярными обладают также и волновыми свойствами. Соотношения, связывающие корпускулярные и волновые свойства частиц, те же, что были установлены ранее для фотонов
E
= h![]() =
= ![]() ω,
ω, ![]() =
=
![]() ,
|p| = h/λ
,
|p| = h/λ![]() /
/![]() ,
,
где
h = 2π
, ω =
2πν, ![]() =
2π
-
длина волны, которую можно сопоставить
с частицей. Волновой вектор
ориентирован
по направлению движения частицы. Прямыми
опытами, подтверждающими идею
корпускулярно-волнового дуализма
частиц, были опыты, выполненные в 1927
году К. Дэвиссоном и Л. Джермером по
дифракции электронов на монокристалле
никеля. Позднее наблюдалась дифракция
и других микрочастиц. Метод дифракции
частиц в настоящее время широко
используется в изучении строения и
свойств вещества.
Экспериментальное подтверждение идеи
корпускулярно-волнового дуализма
привело к пересмотру привычных
представлений о движении частиц и
способа описания частиц. Для классических
материальных точек характерно движение
по определенным траекториям, так, что
их координаты и импульсы в любой момент
времени точно известны. Для квантовых
частиц это утверждение неприемлемо,
так как для квантовой частицы импульс
частицы связан с ее длиной волны, а
говорить о длине волны в данной точке
пространства бессмысленно. Поэтому для
квантовой частицы нельзя одновременно
точно определить значения ее координат
и импульса. Если частица занимает точно
определенное положение в пространстве,
то ее импульс полностью неопределен и
наоборот, частица с определенным
импульсом имеет полностью неопределенную
координату. Неопределенность в значении
координаты частицы Δx и неопределенность
в значении компоненты импульса
частицы Δpx связаны соотношением
неопределенности, установленнымВ. Гейзенбергом в
1927 году
=
2π
-
длина волны, которую можно сопоставить
с частицей. Волновой вектор
ориентирован
по направлению движения частицы. Прямыми
опытами, подтверждающими идею
корпускулярно-волнового дуализма
частиц, были опыты, выполненные в 1927
году К. Дэвиссоном и Л. Джермером по
дифракции электронов на монокристалле
никеля. Позднее наблюдалась дифракция
и других микрочастиц. Метод дифракции
частиц в настоящее время широко
используется в изучении строения и
свойств вещества.
Экспериментальное подтверждение идеи
корпускулярно-волнового дуализма
привело к пересмотру привычных
представлений о движении частиц и
способа описания частиц. Для классических
материальных точек характерно движение
по определенным траекториям, так, что
их координаты и импульсы в любой момент
времени точно известны. Для квантовых
частиц это утверждение неприемлемо,
так как для квантовой частицы импульс
частицы связан с ее длиной волны, а
говорить о длине волны в данной точке
пространства бессмысленно. Поэтому для
квантовой частицы нельзя одновременно
точно определить значения ее координат
и импульса. Если частица занимает точно
определенное положение в пространстве,
то ее импульс полностью неопределен и
наоборот, частица с определенным
импульсом имеет полностью неопределенную
координату. Неопределенность в значении
координаты частицы Δx и неопределенность
в значении компоненты импульса
частицы Δpx связаны соотношением
неопределенности, установленнымВ. Гейзенбергом в
1927 году
Δx·Δpx![]() .
.
Из принципа неопределенности следует, что в области квантовых явлений неправомерна постановка некоторых вопросов, вполне естественных для классической физики. Так, например, не имеет смысла говорить о движении частицы по определенной траектории. Необходим принципиально новый подход к описанию физических систем. Не все физические величины, характеризующие систему, могут быть измерены одновременно. В частности, если время жизни некоторого состояния равно Δt, то неопределенность величины энергии этого состояния ΔE не может быть меньше ΔE/ , т.е.
ΔE·Δt .
Следовательно, нельзя рассчитать траекторию движения электрона в поле ядра, можно лишь оценить вероятность его нахождения в атоме с помощью волновой функции ψ, которая заменяет классическое понятие траектории. Волновая функция ψ характеризует амплитуду волны в зависимости от координат электрона, а ее квадрат ψ2 определяет пространственное распределение электрона в атоме. В наиболее простом варианте волновая функция зависит от трех пространственных координат и дает возможность определить вероятность нахождения электрона в атомном пространстве или его орбиталь. Таким образом, атомная орбиталь (АО) – область атомного пространства, в котором вероятность нахождения электрона наибольшая.
Волновые функции получаются при решении основополагающего соотношения волновой механики – уравнения Шредингера.HΨ=EΨ Точное решение существует для атома водорода или водородоподобных ионов, для многоэлектронных систем используются различные приближения. Поверхность, ограничивающая 90–95 % вероятности нахождения электрона или электронной плотности, называют граничной. Атомная орбиталь и плотность электронного облака имеют одинаковую граничную поверхность (форму) и одинаковую пространственную ориентацию. Атомные орбитали электрона, их энергия и направление в пространстве зависят от четырех параметров – квантовых чисел
В основу математического аппарата квантовой механики положено утверждение, что состояние частицы полностью описывается определенной функцией координат и времени, получившей название волновой функции Ψ(x,y,z) Волновая функция в общем случае комплексная величина и сама по себе физического смысла не имеет. Физический смысл имеет квадрат модуля волновой функции равный плотности вероятности обнаружить частицу в данной точке пространства, т.е. вероятность
обнаружить частицу в элементе объема
Квантовые числа – целые или дробные числа, определяющие возможные дискретные значения физических величин, характеризующих квантовые системы (атомное ядро, атом, молекулу и т.д.) и отдельные элементарные частицы.
Квантовые числа электрона
Квантовое число n – главное. Оно определяет энергию электрона в атоме водорода и одноэлектронных системах (He+, Li2+ и т. д.). В этом случае энергия электрона
|

где n принимает значения от 1 до ∞. Чем меньше n, тем больше энергия взаимодействия электрона с ядром. При n = 1 атом водорода находится в основном состоянии, при n > 1 – в возбужденном.
В многоэлектронных атомах электроны с одинаковыми значениями n образуют слой или уровень, обозначаемый буквами K, L, M, N, O, P и Q. Буква K соответствует первому уровню, L – второму и т. д.
Орбитальное квантовое число l характеризует форму орбиталей и принимает значения от 0 до n – 1. Кроме числовыхl имеет буквенные обозначения
l |
= |
0 |
1 |
2 |
3 |
4 |
… |
l |
= |
s |
p |
d |
f |
g |
… |
Электроны с одинаковым значением l образуют подуровень.
Квантовое
число l определяет квантование
орбитального момента количества движения
электрона ![]() в
сферически симметричном кулоновском
поле ядра.
в
сферически симметричном кулоновском
поле ядра.
Квантовое число ml называют магнитным. Оно определяет пространственное расположение атомной орбитали и принимает целые значения от –l до +l через нуль, то есть 2l + 1 значений. Расположение орбитали характеризуется значением проекции вектора орбитального момента количества движения Mz на какую-либо ось координат (обычноось z):
|
Все вышесказанное можно представить таблицей:
Орбитальное квантовое число
|
||||||||||||||||||
Табл Число орбиталей на энергетических подуровнях |
Орбитали одного подуровня (l = const) имеют одинаковую энергию. Такое состояние называют вырожденным по энергии. Так p-орбиталь – трехкратно, d – пятикратно, а f – семикратно вырождены. Граничные поверхности s-, p-, d-, f- орбиталей показаны на рис
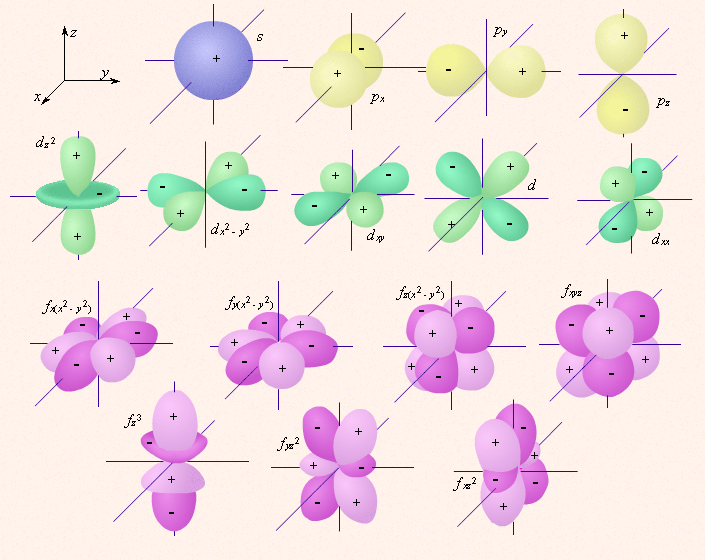
s-Орбитали сферически симметричны для любого n и отличаются друг от друга только размером сферы. Их максимально симметричная форма обусловлена тем, что при l = 0 и μl = 0.
p-Орбитали существуют при n ≥ 2 и l = 1, поэтому возможны три варианта ориентации в пространстве: ml = –1, 0, +1. Все p-орбитали обладают узловой плоскостью, делящей орбиталь на две области, поэтому граничные поверхности имеют форму гантелей, ориентированных в пространстве под углом 90° друг относительно друга. Осями симметрии для них являются координатные оси, которые обозначаются px, py, pz.
d-Орбитали определяются квантовым числом l = 2 (n ≥ 3), при котором ml = –2, –1, 0, +1, +2, то есть характеризуются пятью вариантами ориентации в пространстве. d-Орбитали, ориентированные лопастями по осям координат, обозначаются dz² и dx²–y², а ориентированные лопастями по биссектрисам координатных углов – dxy, dyz, dxz.
Семь f-орбиталей, соответствующих l = 3 (n ≥ 4), изображаются в виде граничных поверхностей, приведенных на рис. 2.1.
Квантовые
числа n, l и ml не полностью
характеризуют состояние электрона в
атоме. Экспериментально установленно,
что электрон имеет еще одно свойство –
спин. Упрощенно спин можно представить
как вращение электрона вокруг собственной
оси. Спиновое квантовое число ms имеет
только два значения ms = ±1/2,
представляющие собой две проекции
углового момента электрона на выделенную
ось. Электроны с разными msобозначаются
стрелками, направленными вверх ![]() и
вниз
и
вниз ![]() .
.
В многоэлектронных атомах, как и в атоме водорода, состояние электрона определяется значениями тех же четырех квантовых чисел, однако в этом случае электрон находится не только в поле ядра, но и в поле других электронов. Поэтому энергия в многоэлектронных атомах определяется не только главным, но и орбитальным квантовым числом, а вернее их суммой: энергия атомных орбиталей возрастает по мере увеличения суммы n + l; при одинаковой сумме сначала заполняется уровень с меньшим n и большим l. Энергия атомных орбиталей возрастает согласно ряду
|
1s < 2s < 2p < 3s < 3p < 4s ≈ 3d < 4p < 5s ≈ 4d < 5p < 6s ≈ 4f ≈ 5d < 6p < 7s ≈ 5f ≈ 6d < 7p. |
|
Итак, четыре квантовых числа описывают состояние электрона в атоме и характеризуют энергию электрона, его спин, форму электронного облака и его ориентацию в пространстве. При переходе атома из одного состояния в другое происходит перестройка электронного облака, то есть изменяются значения квантовых чисел, что сопровождается поглощением или испусканием атомом квантов энергии.
4
Квантово - механические принципы заполнения электронных оболочек атомов Принцип Паули, принцип наименьшей энергии, правило Гунда Примеры составления электронных конфигураций атомов в основном и возбужденном состоянии. «Провал» электрона.
По мере увеличения суммарного числа электронов в атомах (при возрастании зарядов их ядер, или порядковых номеров химических элементов) атомные орбитали заселяются таким образом, что появление электронов на орбитали с более высокой энергией зависит только от главного квантового числа n и не зависит от всех остальных квантовых чисел, в том числе и от l. Физически это означает, что в водородоподобном атоме (в отсутствие межэлектронного отталкивания) орбитальная энергия электрона определяется только пространственной удаленностью зарядовой плотности электрона от ядра и не зависит от особенностей его движения в поле ядра. Поэтому энергетическая последовательность орбиталей в водородоподобном атоме выглядит просто:
1s<2s=2p<3s=3p=3d<4s=4p=4d=4f<5s...
Здесь орбитальная энергия электрона повышается только по мере увеличения главного квантового числа и не меняется при увеличении орбитального квантового числа l; состояния с различными значениями l, но с одним и тем же значением n(например, 3s, Зр, 3d) энергетически эквивалентны, то есть соответствующие атомные орбитали (3s, Зр, 3d) обладают одинаковой энергией и оказываются энергетически вырожденными (не следует путать обсуждаемое вырождение по энергии атомных орбиталей различного типа в гипотетических водородоподобных атомах с энергетическим вырождением атомных орбиталей одного и того же типа, например Зрx, Зру и Зрz в реальных изолированных атомах).
В многоэлектронных атомах в результате эффекта межэлектронных взаимодействий происходит энергетическое расщепление (расхождение) орбиталей различного типа, но с одним и тем же значением главного квантового числа (3s<3p<3d и т. д.). Если бы это расщепление было небольшим и меньшим расщепления по энергии атомных орбиталей под воздействием изменения главного квантового числа n, то энергетическая последовательность атомных орбиталей выглядела бы так:
1s«2s<2p"3s<3p<3d"4s<4p<4d<4f"5s...
В
действительности же расщепление по l,
начиная с n≥З, оказывается большим,
чем расщепление по n. Сложный характер
явления межэлектронных взаимодействий
предопределяет сильную зависимость
орбитальной энергии каждого электрона
уже не только от пространственной
удаленности его зарядовой плотности
от ядра (от главного квантового числа n),
но и от формы его движения в поле ядра
(от орбитального квантового числа l).
Именно межэлектронное взаимодействие
приводит к резко усложнённой (по сравнению
с вышеописанной) энергетической
последовательности заселяющихся
электронами атомных орбиталей. Итак, в
реальных многоэлектронных атомах
картина энергетического распределения
орбиталей оказывается очень сложной.
Строгая квантовомеханическая теория
электронного строения атомов и
экспериментальная спектроскопия
обнаруживают энергетическую
последовательность атомных орбиталей
в следующем виде:
![]()
При́нцип Па́ули (принцип запрета) — один из фундаментальных принципов квантовой механики, согласно которому два и более тождественных фермиона (частиц с полуцелым спином) не могут одновременно находиться в одном квантовом состоянии.
Принцип наименьшей энергии
В атоме каждый электрон располагается так, чтобы его энергия была минимальной (что отвечает наибольшей связи его с ядром).
Энергия электрона в основном определяется главным квантовым числом n и побочным квантовым числом l, поэтому сначала заполняются те подуровни, для которых сумма значений квантовых чисел n и l является наименьшей.
В.М. Клечковский впервые в 1961 г. сформулировал общее положение, гласящее, что электрон занимает в основном состоянии уровень не с минимальным возможным значением n, а с наименьшим значением суммы n + l.
В том случае, когда для двух подуровней суммы значений n и l равны, сначала идет заполнение подуровня с меньшим значением n. Например, на подуровнях Зd, 4р, 5s сумма значений n и l равна 5. В этом случае происходит сначала заполнение подуровней с меньшими значениями n, т.е. Зd - 4р – 5s и т.д. Следовательно, согласно принципу наименьшей энергии во многих случаях электрону энергетически выгоднее занять подуровень «вышележащего» уровня, хотя подуровень "нижележащего" уровня не заполнен
Именно поэтому в четвертом периоде сначала заполняется подуровень 4s и лишь после этого подуровень Зd.
Принцип наименьшей энергии справедлив только для основных состояний атомов. В возбужденных состояниях электроны могут находиться на любых орбиталях атомов, если при этом не нарушается принцип Паули.
ПРАВИЛО ГУНДА |
|
Электроны, заполняющие данный энергетический уровень, стремятся занять максимальное количество орбиталей, располагаясь на каждой по одному, чтобы суммарный спин был максимален. |
|
Это означает, что в каждой из орбиталей подслоя заполняется сначала один электрон, а только после исчерпания незаполненных орбиталей на эту орбиталь добавляется второй электрон. При этом на одной орбитали находятся два электрона с полуцелыми спинами противоположного знака, которые спариваются (образуют двухэлектронное облако) и, в результате, суммарный спин орбитали становится равным нулю.
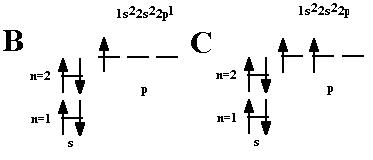
Провал ЭЛЕКТРОНА - отступления от общей для большинства элементов последовательности заполнения электронных оболочек (1s, 2s, 2p, 3s, 3p, 4s, 3d и так далее), связанные с тем, что эти "нарушения правил" обеспечивают атомам некоторых элементов меньшую энергию по сравнению с заполнением электронных оболочек "по правилам,»
Наиболее устойчивы симметрично заполненные подуровни. Или полностью заполненные p6 ( на p подуровне 6 электронов), d10, f 14, или наполовину - р3, d5, f 7. Поэтому для получения более устойчивой и энергетически более выгодной конфигурации происходит проскок или провал электрона на предыдущий подуровень.
5
Периодический закон Д.И. Менделеева. Структура периодической системы. Периодичность свойств элементов. Радиус атомов и ионов Энергия ионизации. Энергия сродства электрону. Электроотрицательность. Их изменение в периодах и группах.
Открытие периодического закона и разработка Периодической системы химических элементов Д. И. Менделеевым явились вершиной развития химии в XIXв. Попытки классифицировать химические элементы имели место и до Менделеева.Менделеев считал, что основной характеристикой элементов являются их атомные веса, и в 1869 г. впервые сформулировал периодический закон:Свойства простых тел, а также формы и свойства соединений элементов находятся в периодической зависимости от величины атомных весов элементов.Несмотря на огромную значимость открытия Менделеева, оно представляло лишь гениальное эмпирическое обобщение фактов, а их физический смысл долго оставался непонятным. Причина заключалась в том, что в XIXв. отсутствовали какие-либо представления о сложном строении атома.Данные о строении ядра и о распределении электронов в атомах позволяют по-новому рассмотреть периодический закон, периодическую систему элементов. На базе современных представлений периодический закон формулируется так:Свойства простых веществ, а также формы и свойства соединений элементов находятся в периодической зависимости от величины заряда ядра атома (порядкового номера).
Структура периодической системы.Из рассмотрения электронных конфигураций атомов наглядно прослеживается периодичность свойств элементов.
Число электронов, находящихся на внешнем уровне в атомах элементов, располагающихся в порядке увеличения порядкового номера, периодически повторяется. Периодическое изменение свойств элементов с увеличением порядкового номера объясняется периодическим изменением числа электронов на их внешних энергетических уровнях. По числу энергетических уровней атома элементы делятся на семь периодов. Первый период состоит из атомов, в которых электронная оболочка состоит из одного уровня, во втором периоде — из двух, в третьем — из трех, в четвертом — из четырех и т.д. Каждый новый период начинается тогда, когда начинает заполняться новый энергетический уровень.
В периодической системе каждый период начинается элементами, атомы которых на внешнем уровне имеют один электрон, — атомами щелочных металлов — и заканчивается элементами, атомы которых на внешнем уровне имеют 2 (в первом периоде) или 8 электронов (во всех последующих) — атомами благородных газов.Далее мы видим, что внешние электронные оболочки сходны у атомов элементов (Li, Na, К, Rb, Сs); (Ве, Mg, Ca, Sr); (F, Cl, Br, I); (Не, Nе, Аr, Кr, Xе) и т.д. Каждая из вышеприведенных групп элементов оказывается в определенной главной подгруппе периодической таблицы: Li, Na, К, Rb, Сs в I группе, F, Сl, Вr, I — в VII и т.д. Именно вследствие сходства строения электронных оболочек атомов сходны их физические и химические свойства.Число главных подгрупп определяется максимальным числом элементов на энергетическом уровне и равно 8. Число переходных элементов (элементов побочных подгрупп) определяется максимальным числом электронов на d-подуровне и равно 10 в каждом из больших периодов.Поскольку в Периодической системе химических элементов одна из побочных подгрупп содержит сразу три переходных элемента, близких по химическим свойствам (так называемые триады Fе-Со-Ni, Ru-Rh-Pd, Os-Ir-Pt), то число побочных подгрупп, так же как и главных, равно 8.По аналогии с переходными элементами, число лантаноидов и актиноидов, вынесенных внизу Периодической системы в виде самостоятельных рядов, равно максимальному числу электронов на f-подуровне, т.е. 14.Таким образом, строгая периодичность расположения элементов в периодической системе химических элементов Д. И. Менделеева полностью объясняется последовательным характером заполнением энергетических уровней.
Периодические свойства элементов
Периодичность свойств атомов элементов можно проиллюстрировать на самых разных их характеристиках. Перечислим важнейшие из них: радиус атома и атомный объем; потенциал ионизации; сродство к электрону; электроотрицательность атома (рис); степени окисления; физические свойства соединений (плотность, температуры плавления и кипения). Потенциал (энергия) ионизации I - энергия, необходимая для отрыва наиболее слабо связанного электрона от атома: X → Х+ + е. Наименьшие потенциалы ионизации - у щелочных металлов, наибольшие - у инертных газов. Сродство к электрону Е - энергия, которая выделяется при присоединении электрона к атому: X + е → X-. Наибольшее сродство к электрону - у галогенов, наименьшее - у металлов.
Радиусы атомов и ионов Вследствие волнового характера движения электрона атом не имеет строго определенных границ. Поэтому измерить абсолютные размеры атомов невозможно. За радиус свободного атома можно принять теоретически рассчитанное положение главного максимума плотности внешних электронных облаков. Это так называемый орбитальный радиус. Из-за различного состояния атомов в молекуле или кристалле не может быть единого типа атомных радиусов. Радиус связанного атома можно считать либо ионным (кристаллическим), либо атомным.
Атомные радиусы разделяют на металлические, которые мы находим в металлах, сплавах или интерметаллических (?) соединениях, и ковалентные, характерные для неметаллов и вообще для ковалентных молекул.
Ковалентные радиусы в свою очередь подразделяют на тетраэдрические, октаэдрическиеи т.д. Безусловно нужно различать радиусы при одинарной, двойной и тройной связях. Существуют ещё атомные радиусы по Брэггу–Слейтеру и орбитальные радиусы.
Ван-дер-ваальсовы радиусы следует рассматривать как радиусы несвязанных атомов. Их находят по межатомным расстояниям в твердом теле или жидкости, где атомы находятся в непосредственной близости друг от друга, но не связаны между собой ионной, ковалентной или металлической связью.
Ковалентные радиусы
 За
ковалентный радиус атома при одинарной
связи принимают половину расстояния
между ядрами 2х одинаковых атомов,
связанных ковалентной связью.
За
ковалентный радиус атома при одинарной
связи принимают половину расстояния
между ядрами 2х одинаковых атомов,
связанных ковалентной связью.
Ионные радиусы
Измерить межъядерное расстояние в молекуле несложно, но решить какая часть этого расстояния приходится на долю катиона, а какая на долю аниона, далеко не просто.
|
|
|
|
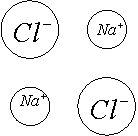
С помощью карты электронной плотности заряда были определены радиусы для катионов и анионов.
По
Брэггу (1920г) межъядерное расстояние в
кристалле можно рассматривать как сумму
радиусов. Ионный радиус отличаются от
атомных (Это определил Слейтер) на
![]() А°,
определив радиус у 1200 соединений.
А°,
определив радиус у 1200 соединений.
Атомные радиусы нужно применять в том случае, когда атомы соединены ковалентной или металлической связью. Это связывание осуществляется, если атомы приближаются друг к другу на расстояние, когда каждый атомный радиус примерно равен радиусу максимума радиальной плотности заряда.
Ионные радиусы используются в том случае, когда внешний электрон полностью удален с внешней орбитали электроотрицательного атома.
Периодичность атомных и ионных R. Для групп непероходных элементов атомные и ионные радиусы увеличиваются с увеличением порядкового номера (Z). Наибольшее увеличение имеет место для двух наиболее легких членов группы (Li и Be) и наименьшее для двух наиболее тяжелых членов группы. Но при изменении атомных и ионных радиусов по периодам они в общем уменьшаются при увеличении заряда ядра (Z). Наибольшее уменьшение наблюдается у элементов малых периодов, т.к. у них происходит заполнение внешнего электронного слоя. В больших периодах в пределах семейств d- и f- элементов наблюдается более плавное уменьшение радиусов. Это уменьшение называют соответственно d и f сжатием, (т.к. эти элементы имеют электроны, обладающие малой экранирующей способностью). В периодах слева направо радиусы атомов меняются незначительно. С увеличением заряда ядра атомов наблюдается постепенное изменение свойств от металлических к типично неметаллическим, что связано с увеличением числа электронов на внешнем энергетическом уровне. Первые три периода содержат только s и р элементы. Четвертый и последующие периоды включают в свой состав также элементы, у которых происходит заполнение d и f подуровней соответствующих внутренних энергетических уровней. При этом f элементы объединяются в семейства, называемые лантаноидами (4 f-элементы) и актиноидами 5 f-элементы. В вертикальных колонках, называемых группами, объединены элементы, имеющие сходное электронное строение внешнего энергетического уровня при различных значениях главного квантового числа и поэтому проявляют сходные химические свойства.
Энергия ионизации
Энергия ионизации — разновидность энергии связи или, как её иногда называют, первый ионизационный потенциал (I1), представляет собой наименьшую энергию, необходимую для удаления электрона от свободного атома в его низшем энергетическом (основном) состоянии на бесконечность.
Энергия ионизации является одной из главных характеристик атома от которой в значительной степени зависят природа и прочность образуемых атомом химических связей. От энергии ионизации атома существенно зависят также восстановительные свойства соответствующего простого вещества.
Для многоэлектронного атома существуют также понятия второго, третьего и т. д. ионизационных потенциалов, представляющих собой энергию удаления электрона от его свободных невозбуждённых катионов с зарядами +1, +2 и т. д. Эти ионизационные потенциалы, как правило, менее важны для характеристики химического элемента.
Энергия ионизации всегда имеет эндоэнергетическое значение (это понятно, так как чтобы оторвать электрон от атома, требуется приложить энергию, самопроизвольно это произойти не может).
На энергию ионизации атома наиболее существенное влияние оказывают следующие факторы:
эффективный заряд ядра, являющийся функцией числа электронов в атоме, экранирующих ядро и расположенных на более глубоко лежащих внутренних орбиталях;
радиальное расстояние от ядра до максимума зарядовой плотности наружного, наиболее слабо связанного с атомом и покидающего его при ионизации, электрона;
мера проникающей способности этого электрона;
межэлектронное отталкивание среди наружных (валентных) электронов.
На энергию ионизации оказывают влияние также и менее значительные факторы, такие, как квантовомеханическое обменное взаимодействие, спиновая и зарядовая корреляция и др. Энергии ионизации возрастает в периоде по мере увеличения порядкового номера элемента. Наименьшее ее значение имеют щелочные металлы, находящиеся в начале периода. Наибольшее значение энергии ионизации характерно для инертных газов, находящихся в конце периода. В группе элементов энергия ионизации уменьшается с повышением порядкового номера элемента. Это обусловлено увеличением размеров атомов и экранированием внешних электронов внутренними.
Эне́ргией сродства́ а́тома к электро́ну, или просто его сродством к электрону (ε), называют энергию, выделяющуюся в процессе присоединения электрона к свободному атому Э в его основном состоянии с превращением его в отрицательный ион Э− (сродство атома к электрону численно равно, но противоположно по знаку энергии ионизации соответствующего изолированного однозарядного аниона).
Э + e− = Э− + ε
Сродство к электрону выражают в килоджоулях на моль (кДж/моль) или в электронвольтах на атом (эВ/атом).
В отличие от ионизационного потенциала атома, имеющего всегда эндоэнергетическое значение, сродство атома к электрону описывается как экзоэнергетическими, так и эндоэнергетическими значениями
Наибольшим сродством к электрону обладают p-элементы VII группы. Наименьшее сродство к электрону у атомов с конфигурацией s2 (Be, Mg, Zn) и s2p6 (Ne, Ar) или с наполовину заполненными p-орбиталями (N, P, As) В подгруппах сверху вниз сродство к электрону атомов уменьшается, но не всегда монотонно. Вследствие экспериментальных трудностей значение сродства к электрону известны не для всех атомов.
Эле́ктроотрица́тельность (χ) — фундаментальное химическое свойство атома, количественная характеристика способности атома в молекуле смещать к себе общие электронные пары. Современное понятие об электроотрицательности атомов было введено американским химиком Л. Полингом. Он использовал понятие электроотрицательности для объяснения того факта, что энергия гетероатомной связи A—B (A, B — символы любых химических элементов) в общем случае больше среднего геометрического значения гомоатомных связей A—A и B—B.
В настоящее время для определения электроотрицательностей атомов существует много различных методов, результаты которых хорошо согласуются друг с другом, за исключением относительно небольших различий, и во всяком случае внутренне непротиворечивы. В периоде электроотрицательность возрастает с увеличением порядкового номера элемента (слева направо), а в группе, как правило, убывает по мере увеличения заряда ядра (сверху вниз). Таким образом , наименьшее значение электроотрицательности имеют s-элементы 1 группы, а наибольшее р-элементы 6 и 7 групп.
6
Ковалентная связь. Основные положения метода валентных связей. Примеры образования ковалентных молекул Основное характеристик ковалентной связи. (энергия и длина связи, насыщаемость, направленность, кратность). Типы ковалентной связи по способу перекрывания орбиталей. Основные типы гибридизации орбиталей.
Ковалентная связь (атомная связь, гомеополярная связь) — химическая связь, образованная перекрытием (обобществлением) пары валентных электронных облаков. Обеспечивающие связь электронные облака (электроны) называются общей электронной парой Термин ковалентная связь был впервые введён лауреатом Нобелевской премииИрвингом Ленгмюром в 1919 году. Этот термин относился к химической связи, обусловленной совместным обладанием электронами, в отличие отметаллической связи, в которой электроны были свободными, или от ионной связи, в которой один из атомов отдавал электрон и становился катионом, а другой атом принимал электрон и становился анионом.
Ковалентная связь - это связь, возникающая между атомами за счет образования общих электронных пар. В основе ее также лежит представление о приобретении атомами энергетически выгодной и устойчивой электронной конфигурации из 8 электронов (для атома водорода из 2). Такую конфигурацию атомы получают не путем отдачи или присоединения электронов как в ионной связи, а посредством образования общих электронных пар. Механизм образования такой связи может быть обменный или донорно-акцепторный. К обменному механизму относят случаи, когда в образовании электронной пары от каждого атома участвует по одному электрону. Например водород: Н2 Н. +Н. →Н:Н или Н-Н. Связь возникает благодаря образованию общей электронной пары за счет объединения неспаренных электронов. У каждого атома есть по одному s –электрону. Атомы Н равноценны и пары одинаково принадлежат обоим атомам. По этому же принципу происходит образование общих электронных пар (перекрывание р-электронных облаков) при образовании молекулы Сl2. При образовании молекулы N2 Образуются 3 общие электронные пары. Перекрываются р-орбитали. Связь называется неполярная. При образовании молекулы хлороводорода перекрывается орбиталь s-электрона водорода и орбиталь р-электрона хлора Н-Сl. Связывающая электронная пара смещена к атому хлора, в результате чего образуется диполь, который измеряется дипольным моментом. Связь называется полярная. По донорно-акцепторному механизму происходит образование иона аммония. Донор (азот) имеет электронную пару, акцептор – (Н+) свободную орбиталь, которую пара электронная азота может занять. В ионе аммония три связи азота с водородом образованы по обменному механизму, а одна по донорно-акцепторному. Все 4 связи равноценны.
Ковалентные связи классифицируют не только по механизму образования общих электронных пар, соединяющих атомы, но и по способу перекрывания электронных орбиталей , по числу общих пар, а также по смещению их. По способу перекрывания – σ (сигма s- s, s-р, р-р) π (р-р гантели перекрываются двумя местами). В молекуле азота между атомами существуют одна σ-связь и две π-связи, которые находятся в двух взаимно перпендикулярных плоскостях. По числу общих электронных пар различают: одинарные Н2, НСl; двойные С2Н4, СО2; тройные N2. По степени смещенности: полярные и неполярные. Связь между атомами с одинаковой электроотрицательностью – неполярная, с разной – полярная. Исследования ученых позволили сделать вывод, что химическая связь в молекуле водорода осуществляется путем образования пары электронов с противоположно направленными спинами. Каждый электрон занимает место в квантовых ячейках обоих атомов, т.е. движется в силовом поле, образованном двумя силовыми центрами – ядрами атомов водорода. Это представление о механизме образования химической связи было развито учеными Гейтлером и Лондоном на примере водорода.это было распространено и на более сложные молекулы. Разработанная на этой основе теория образования химической связи получила название метода валентных связей. Метод ВС дал теоретическое объяснение важнейших свойств ковалентной связи, позволил понять строение большого числа молекул. Хотя этот метод не оказался универсальным и в ряде случаев не в состоянии правильно описать структуру и свойства молекул – все же он сыграл большую роль в разработке квантово-механической теории химической связи и не потерял своего значение до настоящего времени. В основе метода ВС лежат следующие положения: - ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам. -ковалентная связь тем прочнее, чем в большей степени перекрываются взаимодействующие электронные облака. Геометрическая форма s –орбитали сферическая, от центра к краям размазанная (более плотная у ядра, и менее- на краях). Орбитали р-электронов представляют собой гантели, направленные вдоль осей координат. Облака d –электронов имеют более сложную форму.
Энергия связи
Энергия связи – это энергия, которая выделяется при образовании молекулы из одиночных атомов. Энергия связи отличается от ΔHобр. Теплота образования – это энергия, которая выделяется или поглощается при образовании молекул из простых веществ. Так:
N2 + O2 → 2NO + 677,8 кДж/моль – ∆Hобр.
N + O → NO - 89,96 кДж/моль – Е св.
Для двухатомных молекул энергия связей равна энергии диссоциации, взятой с обратным знаком: например в молекуле F2 энергия связи между атомами F-F равна - 150,6 кДж/моль.
Для многоатомных молекул с одним типом связи, например, для молекул АВn, средняя энергия связи равна 1/n части полной энергии образования соединения из атомов. Так, энергия образования СН4 = -1661,1 кДж/моль. Так как в молекуле СН4 четыре связи, то энергия одной связи С – Н равна 415,3 кДж/моль. Исследование большого числа известных в настоящее время данных по энергиям связи показывает, что энергия связи между конкретной парой атомов часто оказывается величиной постоянной при условии, что остальная часть молекулы изменяется незначительно. Так, в насыщенных углеводородах Есв (C – Н) = 415,3 кДж/моль, Есв (C – С) = 331,8 кДж/моль.
Энергии связей в молекулах, состоящих из одинаковых атомов, уменьшаются по группам сверху вниз Если в молекуле соединяются более двух различных атомов, то средняя энергия связи не совпадает с величиной энергии диссоциации молекулы. Если в молекуле представлены различные типы связи, то каждому из них можно приближенно приписать определенное значение Е. Это позволяет оценить энергию образования молекулы из атомов. По периоду энергии связей растут. В этом же направлении возрастает и сродство к электрону
Длина связи
Длина
связи – это расстояние между ядрами
взаимодействующих атомов. Ориентировочно
оценить длину связи можно, исходя из
атомных или ионных радиусов, или из
результатов определения размеров
молекул с помощью числа Авогадро. Так,
объем, приходящийся на одну молекулу
воды: ![]() ,
отсюда
,
отсюда ![]()
С помощью различных методов физико-химических исследований (например электронографии) определяют d более точно. Исследование длин связей показало, что для данной пары атомов в различных (сходных) соединениях длина связи остается величиной постоянной.
Из длин связей между атомами в молекуле можно вычислить ковалентные радиусы атомов. Если рассмотреть гомоядерные двухатомные молекулы с простой связью, такие как F2 или Сl2, атомам F и Сl можно приписать ковалентные радиусы простых связей, равные половине межъядерного расстояния в соответствующих молекулах. Для элементов, которые не могут образовать двухатомные молекулы с простыми связями, используют другие методы определения радиусов. Также можно получить и радиусы кратных связей , чем выше порядок связи между атомами, тем она короче так же На длину связи в значительной мере влияет и гибридизация
Характерные свойства ковалентной связи — направленность, насыщаемость, полярность, поляризуемость — определяют химические и физические свойства соединений.
Направленность связи обусловлена молекулярным строением вещества и геометрической формы их молекулы. Углы между двумя связями называют валентными.
Насыщаемость — способность атомов образовывать ограниченное число ковалентных связей. Количество связей, образуемых атомом, ограничено числом его внешних атомных орбиталей.
Полярность связи обусловлена неравномерным распределением электронной плотности вследствие различий в электроотрицательностях атомов. По этому признаку ковалентные связи подразделяются на неполярные и полярные.
Поляризуемость связи выражается в смещении электронов связи под влиянием внешнего электрического поля, в том числе и другой реагирующей частицы. Поляризуемость определяется подвижностью электронов. Полярность и поляризуемость ковалентных связей определяет реакционную способность молекул по отношению к полярным реагентам.
Электроны тем подвижнее, чем дальше они находятся от ядер. Ковалентная связь это когда два атома делятся электронами и держатся вместе.
Ковалентная связь обладает направленностью. Область перекрывания располагается в определенном направлении по отношению к взаимодействующим атомам. Характер распределения электронов по молекулярным орбиталям позволяет объяснить магнитные свойства частиц. Молекулы, суммарный спин которых равен нулю, проявляют диамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются против направления поля. Молекулы, суммарный спин которых отличен от нуля, проявляют парамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются в направлении поля. Таким образом молекула Н2 диамагнитна. Геометрическая форма молекул зависит от направленности химической связи. Ядра атомов молекул имеющих sр-гибридизацию атомных орбиталей расположены в одной плоскости, sр2 –направлены к вершинам треугольника, sр3 – к верщинам тетраэдра
Примеры веществ с ковалентной связью
Простой ковалентной связью соединены атомы в молекулах простых газов (Н2, Cl2 и др.) и соединений (Н2О, NH3, CH4, СО2, HCl и др.). Соединения с донорно-акцепторной связью — аммония NH4+, тетрафторборат анион BF4− и др. Соединения с семиполярной связью — закись азота N2O, O−-PCl3+.
Кристаллы с ковалентной связью диэлектрики или полупроводники. Типичными примерами атомных кристаллов (атомы в которых соединены между собой ковалентными (атомными) связями могут служить алмаз, германий и кремний.
Единственным известным человеку веществом с примером ковалентной связи между металлом и углеродом является цианокобаламин, известный как витамин B12.
В настоящее время принято изображать электронные пары (то есть химические связи) между атомами черточками. Каждая черточка – это поделенная пара электронов. В этом случае уже знакомые нам молекулы выглядят так:

Формулы с черточками между атомами называются структурными формулами. Чаще в структурных формулах не изображают неподеленные пары электронов, но в ряде случаев (мы столкнемся с ними при обсуждении донорно-акцепторных связей) неподеленные пары играют важную роль.
Структурные формулы очень хороши для изображения молекул: они четко показывают – как атомы связаны между собой, в каком порядке, какими связями.
Связывающая пара электронов в формулах Льюиса – то же самое, что одна черточка в структурных формулах.
Двойные и тройные связи имеют общее название – кратные связи. О молекуле азота говорят, что она имеет порядок связи, равный трем. В молекуле кислорода порядок связи равен двум. Порядок связи в молекулах водорода и хлора – один. У водорода и хлора уже не кратная, а простая связь.
Порядок связи – это число обобществленных поделенных пар между двумя связанными атомами. Порядок связи выше трех не встречается.
Относительную прочность связей можно оценить по энергии, которая необходима для разрыва связей между атомами азота в разных соединениях. Эта энергия дается для одинакового числа молекул таких соединений. Чем выше кратность связи, тем она короче и прочнее.
Чем выше порядок связи, тем прочнее связаны между собой атомы и тем короче сама связь.
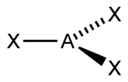
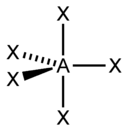
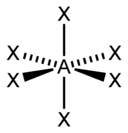
Тип гибридизации |
Число гибридных орбиталей |
Геометрия |
Структура |
Примеры |
sp |
2 |
Линейная |
|
BeF2, CO2, NO2+ |
sp2 |
3 |
Треугольная |
|
BF3, NO3-, CO32- |
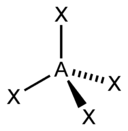
sp3 |
4 |
Тетраэдрическая |
|
CH4, ClO4-, SO42-, NH4+ |
dsp2 |
4 |
Плоскоквадратная |
|
Ni(CO)4, XeF4 |
sp3d |
5 |
Гексаэдрическая |
|
PCl5, AsF5 |
sp3d2 |
6 |
Октаэдрическая |
|
SF6, Fe(CN)63-, CoF63- |

7
Водородная связь. Особенности водородной связи. Межмолекулярное взаимодействие (ориентационное, индукционное, дисперсионное). Примеры. Агрегатное состояние веществ. Типы кристаллических решетох ( атомная, молекулярная, ионная, металлическая).
Водородная связь — форма ассоциации междуэлектроотрицательным атомом и атомом водорода H, связанным ковалентно с другим электроотрицательным атомом. В качестве электроотрицательных атомов могут выступать N, O или F.
Водородные связи могут быть межмолекулярными или внутримолекулярными.
Часто водородную связь рассматривают как электростатическое взаимодействие, усиленное небольшим размером водорода, которое разрешает близость взаимодействующих диполей. Тогда об этом говорят как о разновидности донорно-акцепторной связи, невалентном взаимодействии между атомом водорода H, ковалентно связанным с атомом A группы A-H молекулыRA-H и электроотрицательным атомом B другой молекулы (или функциональной группы той же молекулы) BR'. Результатом таких взаимодействий являются комплексы RA-H•••BR' различной степени стабильности, в которых атом водорода выступает в роли «моста», связывающего фрагменты RA и BR'.
Особенностями водородной связи, по которым её выделяют в отдельный вид, является её не очень высокая прочность, её распространенность и важность, особенно в органических соединениях, а также некоторые побочные эффекты, связанные с малыми размерами и отсутствием дополнительных электронов у водорода.
В настоящее время в рамках теории молекулярных орбиталей водородная связь рассматривается как частный случай ковалентной с делокализацией электронной плотности по цепи атомов и образованием трёхцентровых четырёхэлектронных связей (например, -H•••[F-H•••F]-).
Межмолекулярная и внутримолекулярная водородная связь
Водородные связи обнаружены во многих химических соединениях. Они возникают, как правило, между атомами фтора, азота и кислорода (наиболее электроотрицательные элементы), реже - при участии атомов хлора, серы и других неметаллов. Прочные водородные связи образуются в таких жидких веществах, как вода, фтороводород, кислородсодержащие неорганические кислоты, карбоновые кислоты, фенолы, спирты, аммиак, амины. При кристаллизации водородные связи в этих веществах обычно сохраняются. Поэтому их кристаллические структуры имеют вид цепей (метанол), плоских двухмерных слоев (борная кислота), пространственных трехмерных сеток (лед).
Если водородная связь объединяет части одной молекулы, то говорят о внутримолекулярной водородной связи. Это особенно характерно для многих органических соединений (рис. 42). Если же водородная связь образуется между атомом водорода одной молекулы и атомом неметалла другой молекулы (межмолекулярная водородная связь), то молекулы образуют довольно прочные пары, цепочки, кольца. Так, муравьиная кислота и в жидком и в газообразном состоянии существует в виде димеров:
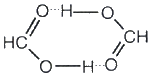
а газообразный фтороводород содержат полимерные молекулы, включающие до четырех частиц HF. Прочные связи между молекулами можно найти в воде, жидком аммиаке, спиртах. Необходимые для образования водородных связей атомы кислорода и азота содержат все углеводы, белки, нуклеиновые кислоты. Известно, например, что глюкоза, фруктоза и сахароза прекрасно растворимы в воде. Не последнюю роль в этом играют водородные связи, образующиеся в растворе между молекулами воды и многочисленными OH-группами углеводов.
Межмолекулярное взаимодействие - взаимодействие молекул между собой, не приводящее к разрыву или образованию новых химических связей. В их основе, как и в основе химической связи, лежат электрические взаимодействия.
Силы Ван-дер-Ваальса включают все виды межмолекулярного притяжения и отталкивания. Они получили название в честь Я.Д. Ван-дер-Ваальса, который первым принял во внимание межмолекулярные взаимодействия для объяснения свойств реальных газов и жидкостей. Эти силы определяют отличие реальных газов от идеальных, существование жидкостей и молекулярных кристаллов. От них зависят многие структурные, спектральные и другие свойства веществ.
Основу ван-дер-ваальсовых сил составляют кулоновские силы взаимодействия между электронами и ядрами одной молекулы и ядрами и электронами другой. На определенном расстоянии между молекулами силы притяжения и отталкивания уравновешивают друг друга, и образуется устойчивая система.
Ван-дер-ваальсовы силы заметно уступают химическому связыванию. Например, силы, удерживающие атомы хлора в молекуле хлора почти в десять раз больше, чем силы, связывающие молекулы Cl2 между собой. Но без этого слабого межмолекулярного притяжения нельзя получить жидкий и твердый хлор.
Ориентационное взаимодействие
Полярные молекулы, в которых центры тяжести положительного и отрицательного зарядов не совпадают, например HCl, H2O, NH3, ориентируются таким образом, чтобы рядом находились концы с противоположными зарядами. Между ними возникает притяжение.

Для взаимодействия двух диполей энергия притяжения между ними (энергия Кеезома) выражается соотношением:
EК = −2 μ1 μ2 / 4π ε0 r3,
где μ1 и μ2 - дипольные моменты взаимодействующих диполей, r - расстояние между ними. Притяжение диполь-диполь может осуществляться только тогда, когда энергия притяжения превышает тепловую энергию молекул; обычно это имеет место в твердых и жидких веществах. Диполь-дипольное взаимодействие проявляется в полярных жидкостях (вода, фтороводород).
Индукционное взаимодействие
Если рядом с полярная молекула окажется полярная рядом с неполярными, она начнет влиять на них. Поляризация нейтральной частицы под действием внешнего поля (наведение диполя) происходит благодаря наличию у молекул свойства поляризуемости γ. Постоянный диполь может индуцировать дипольное распределение зарядов в неполярной молекуле. Под действием заряженных концов полярной молекулы электронные облака неполярных молекул смещаются в сторону положительного заряда и подальше от отрицательного. Неполярная молекула становится полярной, и молекулы начинают притягиваться друг к другу, только намного слабее, чем две полярные молекулы.
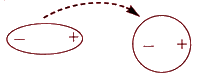
Энергия притяжения между постоянным и наведенным диполем (энергия Дебая) определяется выражением:
EД = −2 μнав2 γ / r6,
где μнав - момент наведенного диполя.
Притяжение постоянного и наведенного диполей обычно очень слабое, поскольку поляризуемость молекул большинства веществ невелика. Оно действует только на очень малых расстояниях между диполями. Этот вид взаимодействия проявляется главным образом в растворах полярных соединений в неполярных растворителях.
Дисперсионное взаимодействие
Между неполярными молекулами также может возникнуть притяжение. Электроны, которые находятся в постоянном движении, на миг могут оказаться окажется сосредоточенными с одной стороны молекулы, то есть неполярная частица станет полярной. Это вызывает перераспределение зарядов в соседних молекулах, и между ними устанавливаются кратковременные связи:
Энергия такого взаимодействия (энергия Лондона) дается соотношением:
EЛ = −2 μмгн2 γ2 / r6,
где μмгн - момент мгновенного диполя. Лондоновские силы притяжения между неполярными частицами (атомами, молекулами) являются весьма короткодействующими. Значения энергии такого притяжения зависят размеров частиц и числа электронов в наведенных диполях. Эти связи очень слабые - самые слабые из всех межмолекулярных взаимодействий. Однако они являются наиболее универсальными, так как возникают между любыми молекулами.
Агрега́тное состоя́ние — состояние вещества, характеризующееся определёнными качественными свойствами: способностью или неспособностью сохранять объём и форму, наличием или отсутствием дальнего и ближнего порядка и другими. Изменение агрегатного состояния может сопровождаться скачкообразным изменением свободной энергии,энтропии, плотности и других основных физических свойств.
Выделяют три основных агрегатных состояния: твёрдое тело, жидкость и газ. Иногда не совсем корректно к агрегатным состояниям причисляют плазму. Существуют и другие агрегатные состояния, например, жидкие кристаллы или конденсат Бозе — Эйнштейна.
Изменения агрегатного состояния это термодинамические процессы, называемые фазовыми переходами. Выделяют следующие их разновидности: из твёрдого в жидкое — плавление; из жидкого в газообразное — испарение и кипение; из твёрдого в газообразное — сублимация; из газообразного в жидкое или твёрдое — конденсация; из жидкого в твёрдое —кристаллизация. Отличительной особенностью является отсутствие резкой границы перехода к плазменному состоянию.
Определения агрегатных состояний не всегда являются строгими. Так, существуют аморфные тела, сохраняющие структуру жидкости и обладающие небольшой текучестью и способностью сохранять форму; жидкие кристаллы текучи, но при этом обладают некоторыми свойствами твёрдых тел, в частности, могут поляризовать проходящее через них электромагнитное излучение.
Для описания различных состояний в физике используется более широкое понятие термодинамической фазы. Явления, описывающие переходы от одной фазы к другой, называют критическими явлениями.
Типы крист решеток
Твердые вещества, как правило, имеют кристаллическое строение. Оно характеризуется правильным расположением частиц в строго определенных точках пространства. При мысленном соединении этих точек пересекающимися прямыми линиями образуется пространственный каркас, который называют кристаллической решеткой.
Точки, в которых размещены частицы, называются узлами кристаллической решетки. В узлах воображаемой решетки могут находиться ионы, атомы или молекулы. Они совершают колебательные движения. С повышением температуры амплитуда колебаний возрастает, что проявляется в тепловом расширении тел.
В зависимости от вида частиц и характера связи между ними различают четыре типа кристаллических решеток: ионные,атомные, молекулярные и металлические.
Ионные кристаллические решетки Виды частиц в узлах решетки: ионы Для веществ с ионной химической связью будет характерна ионная решетка. Ионы-это частицы, имеющие положительный или отрицательный заряд. Напрмер NaCl , Соли, галогениды (IA,IIA),оксиды и гидроксиды типичных металлов. Физ. свойства: Твердые, прочные, нелетучие, хрупкие, тугоплавкие, многие растворимы в воде, расплавы проводят электрический ток Атомные кристаллические решетки В узлах атомной кристаллической решетки находятся отдельные атомы. Ковалентная химическая связь. В данных решетках молекулы отсутствуют. Весь кристалл следует рассматривать как гигантскую молекулу. Примером веществ с таким типом кристаллических решеток могут служить аллотропные модификации углерода: алмаз, графит; а также бор, кремний, красный фосфор, германий. Простые по составу. Атомные кристаллические решетки имеют не только простые, но и сложные. Например, оксид алюминия, оксид кремния. Все эти вещества имеют очень высокие температуры плавления (у алмаза свыше 35000С), прочны и тверды, нелетучи, практически нерастворимы в жидкостях. Металлические кристаллические решетки Металлическая связь. Связь в металлах между положительными ионами посредством обобществленных электронов. общие физические свойства для металлов характерны: блеск, электропроводность, теплопроводность, пластичность. Вещества с металлической связью имеют металлические кристаллические решетки В узлах таких решеток находятся атомы и положительные ионы металлов, а в объеме кристалла свободно перемещаются валентные электроны. Электроны электростатически притягивают положительные ионы металлов. Этим объясняется стабильность решетки. Молекулярные кристаллические решетки Эти вещества являются неметаллами. Простые по составу.Химическая связь внутри молекул ковалентная неполярная.Летучие, легкоплавкие, малорастворимые в воде. в узлах решетки нах. молекулы. молекулярную кристаллическую решетку могут иметь не только твердые простые вещества: благородные газы, H2,O2,N2, I2, O3, белый фосфор Р4, но и сложные: твердая вода, твердые хлороводород и сероводород. Большинство твердых органических соединений имеют молекулярные кристаллические решетки (нафталин, глюкоза, сахар). В узлах решеток находятся неполярные или полярные молекулы. Несмотря на то, что атомы внутри молекул связаны прочными ковалентными связями, между самими молекулами действуют слабые силы межмолекулярного взаимодействия.
8
Первый закон термодинамики. Внутренняя энергия (U)/ Энтальпия (Н) Частные случаи первого закона термодинамики для изохорного и изобарного процессов. Экзотермический и эндотермический эффекты. Закон Гесса и его следствия.
Первый закон термодинамики
На рис. условно изображены энергетические потоки между выделенной термодинамической системой и окружающими телами. Величина Q > 0, если тепловой поток направлен в сторону термодинамической системы. Величина A > 0, если система совершает положительную работу над окружающими телами.
|
Рис Обмен энергией между термодинамической системой и окружающими телами в результате теплообмена и совершаемой работы |
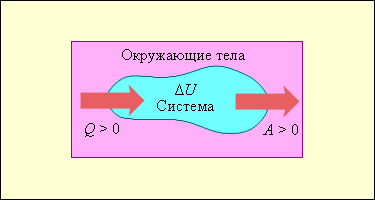
Если система обменивается теплом с окружающими телами и совершает работу (положительную или отрицательную), то изменяется состояние системы, т. е. изменяются ее макроскопические параметры (температура, давление, объем). Так как внутренняя энергия U однозначно определяется макроскопическими параметрами, характеризующими состояние системы, то отсюда следует, что процессы теплообмена и совершения работы сопровождаются изменениемΔU внутренней энергии системы.
Первый закон термодинамики является обобщением закона сохранения и превращения энергии для термодинамической системы. Существует несколько эквивалентных формулировок первого начала термодинамики
В любой изолированной системе запас энергии остаётся постоянным. Это — формулировка Дж. П. Джоуля (1842 г.).
Количество теплоты, полученное системой, идёт на изменение её внутренней энергии и совершение работы против внешних сил
Изменение внутренней энергии системы при переходе её из одного состояния в другое равно сумме работы внешних сил и количества теплоты, переданного системе, то есть, оно зависит только от начального и конечного состояния системы и не зависит от способа, которым осуществляется этот переход. Это определение особенно важно для химической термодинамики (ввиду сложности рассматриваемых процессов). Иными словами, внутренняя энергия является функцией состояния. В циклическом процессе внутренняя энергия не изменяется.
![]()
Изменение полной энергии системы в квазистатическом процессе равно количеству теплоты
 ,
сообщённому системе, в сумме с изменением
энергии, связанной с количеством
вещества
,
сообщённому системе, в сумме с изменением
энергии, связанной с количеством
вещества  при химическом
потенциале
при химическом
потенциале  ,
и работы
,
и работы  ],
совершённой над системой внешними
силами и полями, за вычетом работы
],
совершённой над системой внешними
силами и полями, за вычетом работы  ,
совершённой самой системой против
внешних сил
,
совершённой самой системой против
внешних сил
![]() .
.
Для
элементарного количества теплоты ![]() ,
элементарной работы
,
элементарной работы ![]() и
малого приращения
и
малого приращения ![]() внутренней
энергии первый закон термодинамики
имеет вид:
внутренней
энергии первый закон термодинамики
имеет вид:
![]() .
.
Разделение работы на две части, одна из которых описывает работу, совершённую над системой, а вторая — работу, совершённую самой системой, подчёркивает, что эти работы могут быть совершены силами разной природы вследствие разных источников сил.
Важно
заметить, что
и ![]() являются полными
дифференциалами, а
и
—
нет.
являются полными
дифференциалами, а
и
—
нет.
Согласно этому закону, энергия не может быть создана или уничтожена; она передается от одной системы к другой и превращается из одной формы в другую. Важным следствием первого закона термодинамики является утверждение о невозможности создания машины, способной совершать полезную работу без потребления энергии извне и без каких-либо изменений внутри самой машины. Такая гипотетическая машина получила название вечного двигателя (perpetuum mobile) первого рода. Многочисленные попытки создать такую машину неизменно заканчивались провалом. Любая машина может совершать положительную работу A над внешними телами только за счет получения некоторого количества теплоты Qот окружающих тел или уменьшения ΔU своей внутренней энергии.
Применим первый закон термодинамики к изопроцессам в газах.
В изохорном процессе (V = const) газ работы не совершает, A = 0. Следовательно,
|
Здесь U (T1) и U (T2) – внутренние энергии газа в начальном и конечном состояниях. Внутренняя энергия идеального газа зависит только от температуры (закон Джоуля). При изохорном нагревании тепло поглощается газом (Q > 0), и его внутренняя энергия увеличивается. При охлаждении тепло отдается внешним телам (Q < 0).
В изобарном процессе (p = const) работа, совершаемая газом, выражается соотношением
|
Первый закон термодинамики для изобарного процесса дает:
|
При изобарном расширении Q > 0 – тепло поглощается газом, и газ совершает положительную работу. При изобарном сжатии Q < 0 – тепло отдается внешним телам. В этом случае A < 0. Температура газа при изобарном сжатии уменьшается, T2 < T1; внутренняя энергия убывает, ΔU < 0.
В изотермическом процессе температура газа не изменяется, следовательно, не изменяется и внутренняя энергия газа, ΔU = 0.
Первый закон термодинамики для изотермического процесса выражается соотношением
|
Количество теплоты Q, полученной газом в процессе изотермического расширения, превращается в работу над внешними телами. При изотермическом сжатии работа внешних сил, произведенная над газом, превращается в тепло, которое передается окружающим телам.
Наряду с изохорным, изобарным и изотермическим процессами в термодинамике часто рассматриваются процессы, протекающие в отсутствие теплообмена с окружающими телами. Сосуды с теплонепроницаемыми стенками называются адиабатическими оболочками, а процессы расширения или сжатия газа в таких сосудах называются адиабатическими.
Вну́тренняя эне́ргия тела (обозначается как E или U) — это сумма энергий молекулярных взаимодействий и тепловых движений молекулы. Внутренняя энергия является однозначной функцией состояния системы. Это означает, что всякий раз, когда система оказывается в данном состоянии, её внутренняя энергия принимает присущее этому состоянию значение, независимо от предыстории системы. Следовательно, изменение внутренней энергии при переходе из одного состояния в другое будет всегда равно разности между ее значениями в конечном и начальном состояниях, независимо от пути, по которому совершался переход.
Внутреннюю энергию тела нельзя измерить напрямую. Можно определить только изменение внутренней энергии:
![]()
где
![]() —
подведённое к телу
количество теплоты, измеренное в джоулях
—
подведённое к телу
количество теплоты, измеренное в джоулях
![]() — работа,
совершаемая телом против внешних сил,
измеренная в джоулях
— работа,
совершаемая телом против внешних сил,
измеренная в джоулях
Эта формула является математическим выражением первого начала термодинамики
Для квазистатических процессов выполняется следующее соотношение:
![]()
где
![]() — температура,
измеренная в кельвинах
— температура,
измеренная в кельвинах
![]() — энтропия,
измеренная в джоулях/кельвин
— энтропия,
измеренная в джоулях/кельвин
![]() — давление,
измеренное в паскалях
— давление,
измеренное в паскалях
— химический потенциал
![]() —
количество частиц
в системе
—
количество частиц
в системе
Энтальпи́я, также тепловая функция и теплосодержание — термодинамический потенциал, характеризующий состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных давления, энтропии и числа частиц.
Проще говоря, энтальпия — это та энергия, которая доступна для преобразования в теплоту при определенных температуре и давлении.
Если термомеханическую систему рассматривать как состоящую из макротела (газа) и поршня площадью S с грузом весом Р = pS, уравновешивающего давление газа р внутри сосуда, то такая система называется расширенной.
Энтальпия или энергия расширенной системы Е равна сумме внутренней энергии газа U и потенциальной энергии поршня с грузом Eпот = pSx = pV
![]()
Таким образом, энтальпия в данном состоянии представляет собой сумму внутренней энергии тела и работы, которую необходимо затратить, чтобы тело объёмом V ввести в окружающую среду, имеющую давление р и находящуюся с телом в равновесном состоянии. Энтальпия системы H — аналогично внутренней энергии и другим термодинамическим потенциалам — имеет вполне определенное значение для каждого состояния, т. е. является функцией состояния. Следовательно, в процессе изменения состояния
![]()
Изменение
энтальпии (или Тепловой эффект
химической реакции) не зависит от пути
процесса, определяясь только начальным
и конечным состоянием системы. Если
система каким-либо путём возвращается
в исходное состояние (круговой процесс),
то изменение любого её параметра,
являющегося функцией состояния, равно
нулю, отсюда ![]() ,
или же
,
или же
![]()
Тепловой эффект химической реакции или изменение энтальпии системы вследствие протекания химической реакции — отнесенное к изменению химической переменной количество теплоты, полученное системой, в которой прошла химическая реакция и продукты реакции приняли температуру реагентов.
Чтобы тепловой эффект являлся величиной, зависящей только от характера протекающей химической реакции, необходимо соблюдение следующих условий:
Реакция должна протекать либо при постоянном объёме Qv(изохорный процесс), либо при постоянном давленииQp(изобарный процесс).
В системе не совершается никакой работы, кроме возможной при P = const работы расширения.
Для экзотермических реакций (т.е. реакций, протекающих с выделением тепла) Q > 0, H < 0, а для эндотермических реакций (протекающих с поглощением тепла) Q < 0, H > 0.
Противоположные знаки величин Q и H означают, что энтальпия характеризует тепловые изменения в системе, а теплота – в окружающей среде.
Закон Гесса и его следствия.
Абсолютные значения энтальпии минералов и других веществ определить невозможно (т.к. невозможно определить абсолютное значение внутренней энергии), однако изменение энтальпии вещества (или системы) при переходе его из одного состояния в другое можно измерить с помощью прибора (калориметра) или рассчитать по закону Гесса.
В 1840 г. Г.И. Гесс сформулировал фундаментальный закон термохимии – закон постоянства сумм тепла (закон Гесса):
Тепловой эффект реакции (изменение энтальпии) зависит только от состояния исходных и конечных веществ и не зависит от пути перехода.
Поясним это на примере. Рассмотрим химическую реакцию превращения веществ А и В в вещества C и D. Такая реакция может проходить в одну, две или, например, в четыре стадии:
Согласно закону Гесса, тепловой эффект данной реакции будет один и тот же, независимо от числа и последовательности промежуточных стадий.
Т.е., Нх = Н1 + Н2 = Н3 + Н4 + Н5 + Н6
Следовательно, энтальпия является функцией состояния.
Так как энтальпия является функцией состояния, то для ее определения необходимо выбрать некое (так называемое стандартное) состояние веществ, участвующих в реакции.
Стандартным состоянием вещества называется такое его агрегатное состояние (или кристаллическая модификация), которое наиболее устойчиво при стандартных условиях.
Для кислорода – молекулярный кислород O2 (а не озон O3);
для углерода – графит (а не алмаз);
для серы – ромбическая модификация (а не моноклинная);
для брома – жидкий, а не газообразный Br2.
Важной
характеристикой вещества является
энтальпия его образования: ![]() (f – от английского слова «formation» -
образование).
(f – от английского слова «formation» -
образование).
Энтальпии образования простых веществ приняты равными нулю.
Энтальпия образования сложного вещества – это тепловой эффект реакции образования 1 моль сложного вещества из простых веществ, находящихся в стандартных состояниях. Измеряется в кДж/моль.
Из закона Гесса вытекает несколько следствий, имеющих важное практическое значение:
Первое следствие:
Тепловой
эффект реакции (![]() )
равен разности между суммой стандартных
энтальпий образования конечных веществ
и суммой энтальпий образования исходных
веществ (с учетом стехиометрических
коэффициентов):
)
равен разности между суммой стандартных
энтальпий образования конечных веществ
и суммой энтальпий образования исходных
веществ (с учетом стехиометрических
коэффициентов):
![]() ,
,
где
![]() .
.
Второе следствие:
Если какую-либо реакцию можно представить как совокупность нескольких стадий, то тепловой эффект этой реакции будет равен сумме тепловых эффектов данных стадий.
9
Второй закон термодинамики. Энтропия (S) - мера неупорядоченности системы. Критерий направленности процессов в изолированной системе Свободная энергия Гиббса (G). Критерий направленности процессов в неизолированной системе.
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая, что коэффициент полезного действия не может равняться единице, поскольку для кругового процесса температура холодильника не может равняться абсолютному нулю.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Существуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему»[1] (такой процесс называется процессом Клаузиуса).
Постулат Томсона (Кельвина): «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
Эквивалентность
этих формулировок легко показать. В
самом деле, допустим, что постулат
Клаузиуса неверен, то есть существует
процесс, единственным результатом
которого была бы передача тепла от более
холодного тела к более горячему. Тогда
возьмем два тела с различной температурой
(нагреватель и холодильник) и проведем
несколько циклов тепловой машины,
забрав тепло ![]() у
нагревателя, отдав
у
нагревателя, отдав ![]() холодильнику
и совершив при этом работу
холодильнику
и совершив при этом работу ![]() .
После этого воспользуемся процессом
Клаузиуса и вернем тепло
от
холодильника нагревателю. В результате
получается, что мы совершили работу
только за счет отъёма теплоты от
нагревателя, то есть постулат Томсона
тоже неверен.
.
После этого воспользуемся процессом
Клаузиуса и вернем тепло
от
холодильника нагревателю. В результате
получается, что мы совершили работу
только за счет отъёма теплоты от
нагревателя, то есть постулат Томсона
тоже неверен.
С другой стороны, предположим, что неверен постулат Томсона. Тогда можно отнять часть тепла у более холодного тела и превратить в механическую работу. Эту работу можно превратить в тепло, например, с помощью трения, нагрев более горячее тело. Значит, из неверности постулата Томсона следует неверность постулата Клаузиуса.
Таким образом, постулаты Клаузиуса и Томсона эквивалентны.
Другая формулировка второго начала термодинамики основывается на понятии энтропии:
«Энтропия изолированной системы не может уменьшаться» (закон неубывания энтропии).
Такая формулировка основывается на представлении об энтропии как о функции состояния системы, что также должно быть постулировано.
Второе начало термодинамики в аксиоматической формулировке Рудольфа Юлиуса Клаузиуса (R. J. Clausius, 1865) имеет следующий вид:
Для любой квазиравновесной термодинамической системы существует однозначная функция термодинамического состояния
 ,
называемая энтропией, такая, что ее
полный дифференциал
,
называемая энтропией, такая, что ее
полный дифференциал 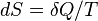 .
.
В состоянии с максимальной энтропией макроскопические необратимые процессы (а процесс передачи тепла всегда является необратимым из-за постулата Клаузиуса) невозможны.
Ограничения
С точки зрения статистической физики второе начало термодинамики имеет статистический характер: оно справедливо для наиболее вероятного поведения системы. Существование флуктуаций препятствует точному его выполнению, однако вероятность сколь-нибудь значительного нарушения крайне мала.
Энтропи́я (от др.-греч. ἐντροπία - поворот, превращение) — в естественных науках мера беспорядка системы, состоящей из многих элементов. В частности, в статистической физике — мера вероятности осуществления какого-либо макроскопического состояния;
Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
![]() ,
,
где ![]() —
приращение энтропии;
—
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса;
—
приращение энтропии;
—
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса;
Энтропия как критерий равновесия и направленности процесса (в изолированных системах).
∆S > 0 - необратимые процессы.
∆S = 0 - обратимый процесс, система близка к равновесному состоянию. На изолированную систему не действуют внешние силы и нет обмена массы, энергия постоянна U=const; V=const; n1=const … nk=const.
1) (δS)U,V,Ni = 0 Признаком того, что функция имеет максимум; вторая производная отрицательная.
2) (δ2S)U,V,Ni < 0
1 и 2 - условие равновесия.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, илитермодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.
Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж)
Классическим определением энергии Гиббса является выражение
где ![]() — внутренняя
энергия,
— внутренняя
энергия, ![]() — давление,
— давление, ![]() — объём,
— объём, ![]() —
абсолютная температура,
—
абсолютная температура, ![]() — энтропия.
— энтропия.
Дифференциал энергии Гиббса для системы с постоянным числом частиц, выраженный в собственных переменных — через давление p и температуру T:
![]()
Для системы с переменным числом частиц этот дифференциал записывается так:
![]()
Здесь — химический потенциал, который можно определить как энергию, которую необходимо затратить, чтобы добавить в систему ещё одну частицу.
Связь с термодинамической устойчивостью системы
Покажем, что минимум потенциала Гиббса соответствует устойчивому равновесию термодинамической системы с фиксированными температурой, давлением и числом частиц.
Запишем обобщённое уравнение первого и второго начал термодинамики:
![]()
При ![]() .
.
![]()
![]()
Таким образом в системе при постоянных температуре и давлении энергия Гиббса достигает минимального значения.
Связь с химическим потенциалом
Используя свойства экстенсивности термодинамических потенциалов, математическим следствием которых является соотношение Гиббса-Дюгема, можно показать, что химический потенциал для системы с одним типом частиц есть отношение энергии Гиббса к числу частиц в системе:
![]()
Если
система состоит из частиц нескольких
сортов ![]() с
числом
с
числом ![]() частиц
каждого сорта, то соотношения Гиббса-Дюгема
приводят к выражению
частиц
каждого сорта, то соотношения Гиббса-Дюгема
приводят к выражению
![]()
Химический
потенциал применяется при анализе
систем с переменным числом частиц, а
также при изучении фазовых переходов.
Так, исходя из соотношений Гиббса-Дюгема
и из условий равенства химических
потенциалов ![]() находящихся
в равновесии друг с другом фаз, можно
получить уравнение Клапейрона-Клаузиуса,
определяющее линию сосуществования
двух фаз в координатах
находящихся
в равновесии друг с другом фаз, можно
получить уравнение Клапейрона-Клаузиуса,
определяющее линию сосуществования
двух фаз в координатах ![]() через
термодинамические параметры (удельные
объёмы) фаз и теплоту перехода между
фазами.
через
термодинамические параметры (удельные
объёмы) фаз и теплоту перехода между
фазами.
Энергия Гиббса и направление протекания реакции
В
химических процессах одновременно
действуют два противоположных
фактора — энтропийный (![]() )
и энтальпийный(
)
и энтальпийный(![]() ).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (
).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (![]() ):
):
![]()
Из
этого выражения следует, что ![]() ,
то есть некоторое количество
теплоты расходуется на увеличение
энтропии (
),
эта часть энергии рассеивается в
окружающую среду в виде тепла, её часто
называют связанной энергией. Другая
часть теплоты (
,
то есть некоторое количество
теплоты расходуется на увеличение
энтропии (
),
эта часть энергии рассеивается в
окружающую среду в виде тепла, её часто
называют связанной энергией. Другая
часть теплоты (![]() )
может быть использована для совершения
работы, поэтому энергию Гиббса часто
называют также свободной энергией.
)
может быть использована для совершения
работы, поэтому энергию Гиббса часто
называют также свободной энергией.
Характер
изменения энергии Гиббса позволяет
судить о принципиальной возможности
осуществления процесса. При ![]() процесс
может протекать, при
процесс
может протекать, при ![]() процесс
протекать не может (иными словами, если
энергия Гиббса в исходном состоянии
системы больше, чем в конечном, то процесс
принципиально может протекать, если
наоборот — то не может). Если же
процесс
протекать не может (иными словами, если
энергия Гиббса в исходном состоянии
системы больше, чем в конечном, то процесс
принципиально может протекать, если
наоборот — то не может). Если же ![]() ,
то система находится в состоянии химического
равновесия.
,
то система находится в состоянии химического
равновесия.
Обратите внимание, что речь идёт исключительно о принципиальной возможности протекания реакции. В реальных же условиях реакция может не начинаться и при соблюдении неравенства (по кинетическим причинам).
Существует
полезное соотношение, связывающее
изменение свободной энергии Гиббса
в
ходе химической реакции с её константой
равновесия ![]() :
:
![]()
Вообще говоря, любая реакция может быть рассмотрена как обратимая (даже если на практике она таковой не является). При этом константа равновесия определяется как
![]()
где ![]() — константа
скорости прямой реакции,
— константа
скорости прямой реакции, ![]() —
константа скорости обратной реакции.
—
константа скорости обратной реакции.
10
Скорость химической реакции. Факторы влияющие на скорость химических реакций. Закон действующих масс - основной закон химической кинетики. Константа скорости химической реакции» ее физический смысл.
Количественной характеристикой того, насколько быстро протекает данная реакция, является скорость химической реакции, т. е. скорость расходования реагентов или скорость появления продуктов. При этом безразлично, о каком из участвующих в реакции веществе идет речь, поскольку все они связаны между собой через уравнение реакции. По изменению количества одного из веществ можно судить о соответствующих изменениях количеств всех остальных.
Скоростью
химической реакции (![]() ) называют
изменение количества вещества реагента
или продукта (
) называют
изменение количества вещества реагента
или продукта (![]()
![]() ) за
единицу времени (
) за
единицу времени (![]() ) в
единице объема (V):
) в
единице объема (V):
= /(V• ).
Скорость реакции в данном случае обычно выражается в моль/(л•с).
Приведенное выражение относится к гомогенным химическим реакциям, протекающим в однородной среде, например между газами или в растворе:
2SO2 + O2 = 2SO3,
BаСl2 + Н2SO4 = ВаSО4 + 2НСl.
Гетерогенные химические реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т.п. К гетерогенным реакциям относятся, например, реакции металлов с кислотами:
Fе + 2НСl = FeСl2 + Н2.
В этом случае скоростью реакции называют изменение количества вещества реагента или продукта ( ) за единицу времени ( ) на единице поверхности (S):
= /(S• ).
Скорость гетерогенной реакции выражается в моль/(м2•с).
Чтобы управлять химическими реакциями, важно не только уметь определять их скорости, но и выяснить, какие условия оказывают на них влияние. Раздел химии, изучающий скорость химических реакций и влияние на нее различных факторов, называется химической кинетикой.
Важнейший фактор, определяющий скорость химической реакции, – концентрация.
При повышении концентрации реагирующих веществ скорость реакции, как правило, возрастает. Для того чтобы вступить в реакцию, две химические частицы должны сблизиться, поэтому скорость реакции зависит от числа столкновений между ними. Увеличение числа частиц в данном объеме приводит к более частым столкновениям и к возрастанию скорости реакции.
Для гомогенных реакций повышение концентрации одного или нескольких реагирующих веществ приведет к увеличению скорости реакции. При понижении концентрации наблюдается противоположный эффект. Концентрация веществ в растворе может быть изменена путем добавления или удаления из сферы реакции реагирующих веществ или растворителя. В газах концентрация одного из веществ может быть увеличена путем введения дополнительного количества этого вещества в реакционную смесь. Концентрации всех газообразных веществ можно увеличить одновременно, уменьшая объем, занимаемый смесью. При этом скорость реакции возрастет. Увеличение объема приводит к обратному результату.
Скорость гетерогенных реакций зависит от площади поверхности соприкосновения веществ, т.е. от степени измельчения веществ, полноты смешивания реагентов, а также от состояния кристаллических структур твердых тел. Любые нарушения в кристаллической структуре вызывают увеличение реакционной способности твердых тел, т.к. для разрушения прочной кристаллической структуры требуется дополнительная энергия. Для протекания химической реакции должно произойти столкновение частиц – атомов, молекул или ионов. В результате столкновений происходит перегруппировка атомов и возникают новые химические связи, что приводит к образованию новых веществ. Вероятность столкновения двух частиц достаточно высока, вероятность одновременного столкновения трех частиц существенно меньше. Одновременное столкновение четырех частиц чрезвычайно маловероятно. Поэтому большинство реакций протекает в несколько стадий, на каждой из которых происходит взаимодействие не более трех частиц.
Скорость элементарных реакций прямо пропорциональна произведению молярных концентраций с (с– это количество вещества в единице объема, с = /V) реагентов, взятых в степенях, равных их стехиометрическим коэффициентам (закон действующих масс для скорости химической реакции). Это справедливо лишь для уравнений реакций, отражающих механизмы реальных химических процессов, когда стехиометрические коэффициенты перед формулами реагентов соответствуют числу взаимодействующих частиц.
По числу взаимодействующих в реакции молекул различают реакции мономолекулярные, бимолекулярные и тримолекулярные. Например, диссоциация молекулярного йода на атомы: I2 = 2I – мономолекулярная реакция.
Взаимодействие йода с водородом: I2 + Н2 = 2HI – бимолекулярная реакция. Закон действующих масс для химических реакций разной молекулярности записывается по-разному.
Мономолекулярные реакции:
А = В + С,
= kcA,
где k – константа скорости реакции.
Бимолекулярные реакции:
А + В = С,
= kcAcВ.
Тримолекулярные реакции:
2А + В = С,
= kc2AcВ.
Столкновение химических частиц приводит к химическому взаимодействию лишь в том случае, если сталкивающиеся частицы обладают энергией, превышающей некоторую определенную величину. Рассмотрим взаимодействие газообразных веществ, состоящих из молекул А2 и В2:
А2 + В2 = 2АВ.
В ходе химической реакции происходит перегруппировка атомов, сопровождающаяся разрывом химических связей в исходных веществах и образованием связей в продуктах реакции. При столкновении реагирующих молекул сначала образуется так называемый активированный комплекс, в котором происходит перераспределение электронной плотности, и лишь потом получается конечный продукт реакции:

Энергию, необходимую для перехода веществ в состояние активированного комплекса, называютэнергией активации.
Активность химических веществ проявляется в низкой энергии активации реакций с их участием. Чем ниже энергия активации, тем выше скорость реакции.
Итак, на скорость реакции оказывает влияние природа реагирующих веществ.