

4.2. Теоретические вопросы к занятию.
1. Определить этиологические и патогенетические факторы наиболее распространенных пороков развития у детей.
2. Врожденные пороки развития: врожденные пороки развития, дизрупции, деформации, дисплазии.
3. Определение порока развития.
4. Определение дизрупции.
5. Определение дисплазии.
6. Дисморфология, как наука. Значение для детской хирургии.
7. Определение синдромологии - раздела клинической медицины. Синдромология разных пороков развития у детей.
8. Синдромы: VATER ассоциация, синдром Опица, синдром Эдварда, синдром Дауна, синдром Гольденхара, CHARGE, синдром ТАР, синдром Нунан, синдром Элерса - Данлоса, синдром Беквита-Видемана.
9. Методы диагностики пороков развития.
10. Показания к обсуждению прерывания беременности при пороках развития и патология, которая требует прерывания беременности.
11. Патология, при которой рекомендовано рождение с помощью Кесарева сечения.
12. Пороки развития, при которых показано неотложное переведение ребенка в хирургическое отделение.
13. Противопоказания к оперативному вмешательству.
4.3. Практические задания, которые выполняются на занятии.
1. Собирать анамнез жизни и заболевания у детей с пороками развития.
2. Проводить осмотр больного с пороками развития.
3. Описывать объективный статус и определять основные клинические и рентгенологические симптомы у детей с пороками развития.
4. Методика обследования ребенка и определение малых пороков развития.
5. Обосновывать и составлять план обследования и лечения детей с пороками развития.
6. Анализировать УЗИ при пороках развития у детей.
7. Определять показания и противопоказания к консервативным и оперативным методам лечения детей с пороками развития.
8. Особенности ведения детей в послеоперационном периоде.
9. Определить реабилитационные мероприятия у детей с пороками развития.
Содержание темы.
На основании современных представлений об этиологии и патогенезе врожденных пороков развития в клинике детской хирургии отводится большое внимание дисморфологии и синдромологии, особенностям диагностики разных синдромальних форм хирургических заболеваний.
Клиническими наблюдениями доказано практическое значение синдромального подхода в работе детского хирурга. Он дает возможность точно диагностировать сопутствующие заболевания у ребенка с множественными врожденными пороками, разрешает прогнозировать характерные для каждого синдрома осложнения при проведении оперативных вмешательств, более точно оценивать возможности хирургического и консервативного лечения, дать точный медико-генетический прогноз в семье.
Значительную часть патологии детского возраста составляют врожденные пороки, т.е. заболевания, обусловленные нарушениями развития эмбриона или плода. При этом всегда нарушается морфология, т.е. структура, форма клеток, ткани органел. Эта область медицины определяется как дисморфология. Изменение может возникать только в одной ткани, в одном органе - эти случаи трактуются как изолированные врожденные пороки. Причиной этих пороков является взаимодействие генетических и внешне-средових факторов, которое позволяет обозначать их, как многофакторные заболевания.
Соответственно международной классификации, все врожденные пороки развития подразделяются на 4 группы:
- врожденные пороки развития,
- дизрупции,
- деформации
- дисплазии.
Пороки развития - стойкие морфологические изменения органа, системы или организма, которые выходят за пределы вариаций их строения и возникают внутриутробно в результате нарушений развития зародыша или (намного реже) после рождения ребенка, как следствие нарушения дальнейшего формирования органов.
Частота пороков развития, по разным данным, колеблется от 2,7% до 16,3%, что зависит преимущественно от полноты учета и возраста обследуемых. В популяции частота пороков развития довольно стабильная, тем не менее в перинатальной и ранней детской смертности их удельный вес из года в год возрастает, что связывают главным образом со снижением смертности от внутриутробной асфиксии, родовых травм и инфекций.
Дизрупция - анатомический дефект органа в результате вторичного нарушения эмбрионального дифференцирования при нормальном генотипе (тератогенные дефекты, вызванные внешними по отношению к эмбриону влияниями - внутриутробными инфекциями, радиацией, химическими и медицинскими препаратами, заболеваниями матери)
Деформация - аномальная форма или аномальное положение части тела, вызванное механической причиной в период внутриутробного развития без нарушения эмбрионального дифференцирования (врожденная косолапость, кривошея, врожденная воронкообразная деформация грудной клетки и другое)
Дисплазия - морфологический дефект ткани в результате первичного генетически детерминированного нарушения дифференцирования этой ткани (гемангиома, пигментные неусы, неоплазии и т.д. ).
Аплазия (агенезия) - врожденное отсутствие органа.
Врожденная гипоплазия (гипотрофия) - недоразвитие органа, которое сопровождается дефицитом его массы или уменьшением размеров, которые превышают отклонения от средних показателей для данного возраста.
Врожденная гиперплазия (гипертрофия) - увеличение массы органа или его размеров за счет роста числа и объема клеток.
Макросомия (гигантизм) - увеличение длины тела.
Гетеротопия (дистопия) - наличие клеток или тканей одного органа в другом или в тех зонах того же органа, где их не должно быть в норме. Дистопию тканей нередко называют хористией, а дистопию с опухолеподобным разрастанием - гамартией.
Гетероплазия - нарушение дифференцирования клеток в пределах одной и той же ткани (например, наличие клеток плоского эпителия в дивертикуле Меккеля).
Эктопия - расположение органа в необычном месте.
Атрезия - отсутствие естественного канала или отверстия.
Персистирование - сохранение эмбриональных структур, в норме исчезающих к определенному периоду развития (например, наличие овального окна в межпередсердной перегородке у ребенка, который достиг 1 года).
Дизрафия (арафа) - незарощення эмбриональных щелей (розщелина верхней губы, верхней челюсти и неба, краниорахишизис - не смыкание костей черепа и позвоночника, по обыкновению сопровождается развитием черепно-мозговых и спинномозговых грыж).
Врожденными аномалиями (малыми пороками) чаще называют пороки развития, которые не сопровождаются нарушениями функций органа, например деформации ушных раковин, не искажают больного и не влияют на функции органа слуха.
Малые аномалии развития, которые встречаются на определенных участках:
Голова: аномальный рисунок роста волоса, уплощенный затылок, "бугры" свода черепа
Участок рта и ротовой полости: микрогения, расщепление язычка, аберрантные уздечки предверия рта, гипоплазия эмали зубов, микродентия, аномально растущие зубы.
Орбитальный участок: эпикантные складки, обратный эпикант, монголоидный разрез глаз, антимонголоидный разрез глаз, короткие глазные щели, дистопия внешних углов глаза, гипотелоризм умеренный, птоз легкий, гетерохромия радужек, микрокорнеа.
Ушные раковины: примитивная форма, дарвинов бугорок, аномальная форма завитка: асимметричная, ротированная, уменьшенная, оттопыренная, отсутствие козелка, расщепление мочки, отсутствие мочки, аурикулярные ямки, аурикулярные выступы.
Шея: крылобразная шея, свищи шеи.
Кисти: рудиментарная полидактилия, единая сгибающая складка ладони, аномальная дерматоглифика, рудиментарная полидактилия, клинодактилия мизинцев, укорочение 4-5 пальцев.
Стопы: синдактилия ІІ-ІІІ пальцев стопы, сандалеобразные щели, утолщение ногтей, короткий палец.
Кожные покровы: гемангиомы, гиперпигментация кожи, неусы, депигментация кожи.
Малые аномалии развития у новорожденных могут быть единым или изолированным признаком - с частотой 14%, или множественные (две и больше малые аномалии развития у ребенка) - с частотой до 11%.
У грудного ребенка с наличием трех и более малых аномалий развития существует 90% достоверность врожденного дефекта развития - нужен поиск порока.
У ребенка с тремя и больше малыми аномалиями развития можно диагностировать определенный синдром с вероятностью 40%, поэтому необходима своевременная диагностика пороков развития.
В зависимости от этиологии все пороки развития разделяют на те что возникли в результате:
- генных мутаций (мономутантные пороки развития);
- хромосомных и геномних мутаций (хромосомные синдромы);
- комбинированного влияния генных мутаций и факторов внешней среды по отношению к зародышу (мультифакторные пороки развития);
- тератогенных факторов (группа бластопатий, эмбриопатий и фетопатии).
Комплексными клиническими, морфологическими, генетическими исследованиями показано, что около 20% всех пороков развития представляют мономутантные формы, 9-12% - хромосомные синдромы, до 65% - мультифакторные пороки развития. Таким образом, подавляющее большинство пороков развития обусловлены (или связаны) с изменениями наследственного материала и лишь 2-5% индуктированы непосредственно тератогенными факторами.
В основе мономутантного порока развития лежит мутация одного гена, которая состоялась в половых клетках родителей или более отдаленных родственников больного. Передача мономутантных пороков развития от родителей детям определяется законами наследственности. В зависимости от типа наследования такие пороки развития могут быть доминантными (например, некоторые формы полидактилии, поликистоз почек, синдром Марфана) и рецесивними (например, инфантильный поликистоз почек, синдром Меккеля).
При доминантно-наследственных пороках развития у одного из родителей у ребенка обычно диагностируется аналогичная аномалия. При рецесивним наследовании родители здоровы, но являются носителями измененного гена.
Хромосомные синдромы (хромосомные болезни) - группа пороков развития, индуцированных численными или структурными изменениями хромосом. К нарушениям числа хромосом относятся трисомии, когда есть дополнительные хромосомы, и моносомия, когда одна из хромосом отсутствует. У людей встречается только моносомия X. Основными структурными изменениями хромосом, которые приводят к порокам развития, являются частичные трисомии и частичные моносомии (делеции).
Хромосомные синдромы проявляются множественными, реже системными пороками развития (некоторые случаи или трисомии Х у женщин и дисомии Х у мужчин). У ребенка с каким-нибудь хромосомным синдромом, как правило, наблюдается большое количество пороков развития. Их объединение создает довольно специфический для большинства хромосомных синдромов патологический морфотип. Известны синдромы, обусловленные мутациями практически любой хромосомы. Из них наиболее часто встречаются синдром Дауна, синдром Клайнфелтера, синдром Шерешевського - Тернера, синдромы Патау, Едвардса, синдромы частичных моносомий по 4, 5 и 18- и хромосомам.
Причиной пороков развития у человека являются лишь немногие из большого перечня тератогенных факторов, известных в экспериментальной тератологии. К таковым, в частности, относятся некоторые вирусы (краснухи), возбудители токсоплазмоза, листериоза, воздействие ионизирующего излучения в суммарной дозе на плод более 0,05 Гр за период органогенеза, некоторые лекарственные препараты (талидомид, варфарин, цитостатики, прогестин, этистерон, метилтестостерон), этиловый алкоголь, сахарный диабет.
Патогенез пороков развития (тератогенез) изучен недостаточно, Установлено, что формирование пороков развития происходит в результате нарушения процессов размножения, миграции и дифференцирования клеток, гибели отдельных клеточных масс, замедление их рассасывания, нарушение адгезии тканей.
Остановка или замедление размножения клеток приводит к аплазии или гипоплазии органа, а также к нарушению слияния. Например, тяжелые симметричные расщелины лица образуются в результате нарушения миграции клеток нейроэктодермального гребня в максиллярные отростки. Нарушение дифференциации клеток, возможно в любом периоде эмбриогенеза, обуславливает агенезии органов, их морфологическую и функциональную незрелость, а также персистирование эмбриональных структур.
Избыточная гибель клеток, отмирающих в процессе нормального эмбриогенеза (например, происходящая при рассасывании межпальцевых перепонок), лежит в основе эктродактилии — аплазии средних пальцев кистей или стоп (клешнеобразные кисть и стопа). Задержка физиологического распада клеток (например, при реканализации кишечной трубки и открытие естественных отверстий) может приводить к атрезии, стенозу.
В основе формирования некоторых пороков развития лежат циркуляторные расстройства, обусловленные тромбозом, сдавленнием, кровоизлиянием. Тератогенный эффект инфекций чаще связан с цитолитическим действием.
Формирование большинства пороков развития происходит в первые 8-10 недель беременности. Выделяют два критических периода, на протяжении которых зародыш наиболее чувствителен к действию повреждающих факторов.
Первый из них приходится на конец 1-й — начало 2-й недели беременности. Повреждающее воздействие в этот период в основном приводит к гибели зародыша. Аналогичное воздействие во втором критическом периоде (3—6-я неделя) чаще индуцирует порок развития. С целью установления возможной этиологии порока развития целесообразно время действия предполагаемого фактора сопоставлять не с критическим, а с тератогенетическим терминационным периодом (ТТП).
Пороки развития разделяют по их этиологии, времени возникновения и объекту повреждения, последовательности формирования, распространенности.
Различают первичные пороки развития (обусловленные непосредственным воздействием повреждающего фактора) и вторичные, являющиеся осложнением первичных. По распространенности выделяют изолированные (одиночные, локальные) пороки развития, локализованные в одном органе; системные пороки развития (в пределах одной системы, например хондродисплазия); множественные пороки развития, развивающиеся в органах двух и более систем (например, сочетание расщелины губы с полидактилией).
Изолированные и системные пороки развития классифицируют по анатомическому признаку на пороки развития центральной нервной системы, сердца и сосудов, дыхательной системы и др.
Множественные пороки развития разделяют на синдромы и неклассифицированные комплексы. Под синдромами понимают устойчивые сочетания первичных пороков развития, возникновение которых индуцировано общим этиологическим фактором (мутацией или тератогенным воздействием). В тех случаях, когда комплекс обнаруженных у больного порок развития не укладывается ни в один из известных синдромов, пользуются термином «неклассифицированный комплекс врожденных пороков» или «множественные пороки, неуточненные».
Среди всех пороков развития одно из первых мест занимают аномалии опорно-двигательного аппарата, при этом 3/4 их приходится на пороки развития конечностей.
С развитием методов диагностики в современной медицине и генетике стали описываться много новых синдромальных форм патологии человека. Хорошо известный каталог Мак Кюсика в электронном варианте в сети Іnternet начисляет уже более 5000 нормальных и патологических признаков человека, которые наследуются соответственно законам Менделя, и число этих признаков увеличивается ежемесячно.
Известная Лондонская база данных по синдромологии в данное время начисляет больше 2500 синдромов, и каждый год описывается 10-15 "новых" нозологических форм синдромальной патологии человека и этот процесс определения "новых" синдромов бесконечный. К началу 80- х лет объем информации в этой области стал такой большой и разнообразный, что нужна была унификация современной терминологии, которая касается определения синдромов и подобных форм множественного поражения человеческого организма.
В 1982 г. появляется первая монография М. Соhеn ( Соhеn М.M., Jr The Chіld wіth Multіple Bіrth Defects.-New York, 1982), в которой разрабатываются методологические основы синдромологии, как практической и научной области медицины. Отличие синдромологии, т.е. дисциплины для диагностики и изучения изолированной патологии человека (порок одного органа или системы), наглядно иллюстрирует тот факт, что в классической медицине на протяжении XX века описано всего несколько новых заболеваний (лучевая болезнь, болезнь легионеров, СПИД, болезнь Лайма), тогда, как в синдромологии число нозологических форм значительно больше. Для некоторых форм синдромальной патологии, современная молекулярная генетика разрешила локализовать детерминующие их гены и исследовать продукты генной транскрипции, которые чаще представлены мембранными рецепторами или тканевыми факторами роста. Так, при болезни Гиршпрунга установлены две мутации разных генов, которые позволили выделить два генетических типа этой врожденной патологии.
Информативные морфогенетические варианты, или малые аномалии развития, — это аномальные варианты морфологии отдельных органов или тканей, не имеющие медицинского значения, т. е. не требующие лечения. В клинической генетике и синдромологии малые аномалии развития, особенно когда у ребенка их насчитывается более трех — чрезвычайно важный диагностический признак, свидетельствующий о высокой вероятности серьезных нарушений морфогенеза в виде врожденных пороков развития, требующих специальной диагностики и нередко последующих хирургических вмешательств. Более 70% всех малых аномалий развития располагаются в области головы, шеи и кистей рук. Диагностическая ценность разных малых аномалий развития различна, и принципиально важно то, что практическая значимость этих признаков заключается в диагностике трех аномалий и более. Каждый новорожденный с тремя и более малыми аномалиями развития имеет высокую вероятность (90%) серьезного врожденного порока развития сердца, почек или позвоночника, кроме этого у такого новорожденного существует высокая вероятность (40%) диагностики синдромальной формы патологии. Иногда только наличие одного типа малых аномалий развития информативно для диагностики, например, полителия (добавочные соски) часто свидетельствует о патологии мочевыделительной системы.
Приблизительно у 1% новорожденных существует объединение нескольких малых аномалий развития и врожденных дефектов, из которых в 40% можно диагностировать тот или другой синдром, а в 60% случаев нужно выделение так называемых новых синдромов. Это свидетельствует о сложности диагностики синдромов, количество которых в данное время превышает 1500, причем каждый год в периодической литературе описывается не меньше 10-15 новых нозологических форм. Частота большинства синдромальных форм патологии довольно низкая (І случай на 10 000-100 000 родов), однако в общей структуре заболеваемости удельный вес синдромальних форм значительно больше. Так, например, среди детей с атрезией пищевода частота синдромальних форм патологии достигает 55%, среди детей с аноректальними пороками - 60%, среди детей с врожденными деформациями грудной клетки - 30%. Отдельные синдромы встречаются наиболее часто, что требует умения их диагностики не только генетиком, но и педиатром, детским хирургом. Так, например, среди детей с крипторхизмом и врожденными пороками сердца встречается синдром Нунен, частота которого в общей популяции составляет 1 случай на 2000 детей; среди новорожденных с эмбриональной и пупочной грыжей чаще диагностируется синдром Беквита-Видеманна с частотой не меньше 1 случай на 12000 родов.
Некоторые синдромы хорошо известны в хирургической практике, как чрезвычайно важные, способные вызвать серьезные осложнения. Например, синдром Элерса-Данлоса описан не менее, чем в 500 публикациях и нескольких монографиях. Он имеет важную роль в детской хирургии и сосудистой хирургии. Подозрение на синдромальную патологию будет обосновано у ребенка с двусторонним пороком, например в случаях двустороннего врожденного порока кисти или стопы (полидактилия, врожденная косорукость). Некоторые врожденные пороки развития или малые аномалии развития с высокой вероятностью указывают на синдромальную патологию или определенный врожденный порок. Так, преаксиальная полидактилия (удвоение первого пальца кисти или стопы) с большой степенью вероятности свидетельствует о синдромальной патологии, тогда, как постаксиальная полидактилия (удвоение мизинца кисти или стопы) обычно является изолированным врожденным пороком развития.
Термин "синдром" греческого происхождения и означает "бегущие рядом". В области патологии человека этот термин обозначает симптомокомплекс, т. е. одновременное наличие у больного двух симптомов и более. Если эти симптомы объединены патогенетическим родством, но могут иметь разную причину, то это патогенетические синдромы.
Хорошим примером подобного синдрома может служить гепатоспленомегалия, причиной которой могут быть врожденный порок развития, опухоль или метаболический дефект. Если же эти симптомы или симптомокомплексы обусловлены одной причиной (монокаузальная этиология), то в этом случае термин "синдром" обозначает нозологическую форму заболевания (нозологический синдром) и в этом смысле является синонимом термина "болезнь".
Выявление у новорожденного трех и более малых аномалий развития требует тщательного ультразвукового исследования сердца, головного мозга, почек и органов брюшной полости с целью своевременной диагностики врожденных пороков развития, которые еще не имеют клинических проявлений в этом возрасте. Кроме того, необходима консультация врача-генетика с целью своевременной диагностики определенных синдромов с дальнейшим диспансерным наблюдением.
Согласно практической дисморфологии, при диагностике у ребенка врожденного порока развития, перед хирургом встают следующие вопросы:
- насколько часто этот врожденный порок ассоциируется с другими врожденными пороками, клинически еще не проявляющимися;
- как часто данный врожденный порок является симптомом синдромальной формы патологии;
- какие синдромы наиболее часто встречаются при данном врожденном пороке.
Ответы на эти вопросы являются первым диагностическим этапом практической совместной работы хирурга и синдромолога. Конечной целью этого этапа является диагностика дополнительных врожденных дефектов развития или определенного синдрома у ребенка. При диагностировании синдромальной формы патологии в большинстве случаев становится ясной дальнейшая врачебная тактика в отношении оперативного или консервативного лечения и медико-генетического прогноза в семье больного. Информация о прогнозе для жизни и здоровья при том или ином синдроме имеет чрезвычайно важное значение и является основной целью врачебной работы.
Первый диагностический этап можно проиллюстрировать на примере одного из наиболее частых врожденных дефектов - атрезии пищевода. Наиболее частые ассоциации, связанные с атрезией пищевода, представленны в табл. 1.
Таблица 1.
Сопутствующие врожденные пороки при атрезии пищевода.
Сопутствующие врожденные пороки |
Частота в % |
Врожденные пороки сердца |
29-37 |
Аноректальные пороки развития |
11-17 |
Врожденные пороки желудочно-кишечного такта |
5-13 |
Врожденные пороки мочеполовой системы |
11-28 |
Врожденные пороки опорно-двигательного аппарата |
10-49 |
Приблизительно в 55% всех случаев атрезии пищевода оказываются сопутствующие врожденные пороки развития, наиболее часто, врожденные пороки сердца и опорно-двигательного аппарата(Stеvenson R.E., Hall J.G., 1996). Синдромальные формы атрезии пищевода встречаются с частотой около 20% и представлены 24-мя формами нозологического диагноза. Наиболее частыми синдромальными формами атрезии являются VATER( Vertebral, Аnorectal, Tracheo Esophageal, Renа1) ассоциация, синдром Опица и синдром трисомии по хромосоме 18 (табл. 2).
Таблица 2.
Синдромология различных форм атрезии пищевода.
Синдром |
Основные проявления |
Этиология |
VATER ассоциация |
Вертебральные, аноректальные, трахео-эзофагиальные, ренальные |
Многофакторное наследование |
Синдром Опица |
Гипертелоризм, гипоспадия, дисфагия, порок сердца, незаращение губы и неба. |
Аутосомно - доминантное наследование |
Синдром Эдвардса |
Выступающий затылок, мелкие черты лица, короткая грудина, дуги на пальцах, задержка психомоторного развития |
Трисомия по 18 хромосоме |
Синдром Дауна |
Мышечная гипотония, макроглосия, луноподобное лицо, монголоидный разрез глаз, гипоплазия V пальца |
Трисомия по 20 хромосоме |
Синдром Гольденхара |
Асимметрия/ гипоплазия лица, тугоухость, полупозвонки позвоночника, макростомия, эпибульбарный дермоид, колобомбы века, порок сердца, гипоплазия легких, порок почек, аномалии почек |
Неизвестно |
CHARGE |
Колобомы (Coloboma), порок сердца (Heartrefects), атрезия хоан (Atresіa choane), задержка роста (Retarded groroth), гипоплазия половых органов (Genіtal hypoplasіa), аномалии ушных раковин (Ear anomalіes) |
Многофакторное наследование |
Большинство хромосомных синдромов с патологией аутосом характеризуется врожденными пороками головного мозга, что делает витальный прогноз при этих заболеваниях крайне неблагоприятным, а у выживших детей отмечается умственная отсталость. Однако у детей с VATER ассоциацией и синдромом Опица чаще нормальный интеллект и витальный прогноз зависит только от тяжести множественных врожденных дефектов.
Важно отметить, что диагностика этих форм патологии основывается только на клинической картине и особенностях фенотипа больного ребенка. Так, синдром Опица должен быть заподозрен при наличии всего двух признаков у мальчиков — выраженного гипертелоризма (широко расположенные глаза) и гипоспадии. Только эти признаки позволяют целенаправленно искать патологию пищевода, особенно при наличии в клинической картине симптомов дисфагии или аспирационной пневмонии.
Разные врожденные пороки характеризуются разной частотой сидромальных форм. Чаще всего встречаются синдромальные формы таких пороков развития, как гастрошизис, экстрофия мочевого пузыря и эписпадия. С другой стороны, при аноректальных дефектах частота синдромальных форм достигает 65%.
Более часто синдромальные формы диагностируются у детей с ортопедической патологией - 58%, с аноректальными аномалиями - 62%, а при патологии пищевода этот показатель составляет - 34%.
Практическая ценность синдромального подхода в хирургической клинике заключается в следующем:
- он дает возможность точно диагностировать сопутствующие заболевания у ребенка с множественными дефектами;
разрешает прогнозировать характерные для каждого синдрома осложнения при проведении оперативного лечения, более точно оценить возможности оперативного или консервативного лечения хирургических болезней (сроки и объем хирургического вмешательства, отдаленные результаты лечения),
дать точный медико-генетический прогноз в семье.
Синдром Нунан назван по имени автора, описавшего это заболевание в 1963 г.) — наследственное аутосомно доминантное заболевание, встречающееся с частотой 1 на 1000—2500 населения. Диагностика основывается на сочетании низкорослости, широкой или крыловидной шеи, деформации грудной клетки, врожденных пороков сердца и необычного лицевого фенотипа: лобные бугры, гипертелоризм, ротированные ушные раковины со свисающими завитками. Дети, практически всегда являются первыми пациентами именно хирургов, так как с рождения диагностируются врожденные пороки сердца (клапанный стеноз легочной артерии, дефект межжелудочковой перегородки), крипторхизм у мальчиков в 60% случаев, комбинированная деформация грудной клетки.
Практические рекомендации для хирургов следующие:
- возможно развитие тяжелого хилоторакса/хилоперикарда при повреждении грудного лимфатического протока,
- гипоплазия или аплазия лимфатических сосудов выявляется в 20% новорожденных в виде генирализованного или периферического лимфостаза,
- возможна злокачественная гипертермия при проведении анестезии,
- высока вероятность кровотечения в послеоперационном периоде в результате дефицита фактора ІX (болезнь Виллебрандта или нарушение функции тромбоцитов, которые возможны у 20% новорожденных),
При проведении кардиохирургических вмешательств и повреждении грудного лимфатического протока развивается хилоперикард или хилоторакс; возможна злокачественная гипертермия при проведении анестезиологического обеспечения. Эта вероятность невысокая -1-2%, но, учитывая серьезность данного осложнения, рекомендуется соблюдать осторожность при проведении анестезии и использовании препарата дартролена.
При синдроме Нунан хирург должен провести исследование свертывающей системы крови, даже при планировании небольших по объему хирургических вмешательств.
Синдром ТАР (первые буквы обозначают клинические проявления заболевания — тромбоцитопения, аплазия, радиальная). Это аутосомно-рецессивное заболевание, характеризующееся сочетанием тромбоцитопении и двусторонним отсутствием лучевой кости с сохранением первого пальца кисти. Диагноз заболевания предельно прост, однако клиническая картина может быть довольно разнообразной с гипоплазией локтевой и/или плечевой кости, наличием врожденного порока сердца
В 90% случаев тромбоцитопения диагностируется в первые 4 месяца жизни, в 60-70% случаев отмечаются лейкемоидные реакции с гепатоспленомегалиемий, в 50% случаев - эозинофилия, иногда возникает гемолиз. Все эти гематологические проявления провоцируются многими факторами, важнейшими из которых являются белки коровьего молока, хирургический стресс и инфекции. Особенно опасны эти факторы у детей до 3- х лет жизни.
Хирургическое вмешательство у детей с синдромом ТАР до 5 летнего возраста проводится только по жизненным показаниям. Исключение из пищевого рациона этих больных белков коровьего молока, осторожность и оптимальные сроки проведения хирургической коррекции лучевой косорукости и врожденных пороков сердца, позволяют сохранить жизнь этим пациентам. При невыполнении этих рекомендаций смертность составляет около 60%, в основном от кровоизлияния в мозг или в легкие. При проведении хирургических вмешательств по жизненным показаниям следует заранее заготовить свежую тромбоцитарную массу для переливания, причем она должна быть взята только от одного донора, так как иммунный конфликт — дополнительный фактор для возникновения тромбоцитопении и лейкемоидных реакций
Элерса - Данлоса. Особенности хирургического ведения и осложнения у больных с синдромом Элерса - Данлоса:
• выражена хрупкость сосудистой стенки (возможность разрывов крупных артерий, несостоятельность хирургического шва); • возможность спонтанного разрыва полых органов (кишечник, мочевой пузырь),
• необходимо соблюдать осторожность при проведении лапароскопии; • необходимо соблюдать осторожность при проведении ангиографического исследования (разрыв артерий);
• высока вероятность спонтанного пневмоторакса; • замедленное формирование послеоперационного рубца (сроки снятия швов увеличены в 1,5—2 раза).
Синдром Беквита-Видеманна - доминантный синдром. Минимальные диагностические критерии: большая масса при рождении или постнатальное опережение физического развития, дефекты закрытия передней стенки живота (эмбриональная грыжа, пупочна грыжа, диастаз прямых мышц живота), висцеромегалия (нефромегалия, гепатомегалия, спленомегалия), макроглосия, необыкновенный вид ребенка (гемангиома кожи лба, "насечки" на ушной раковине). Диагноз этого заболевания следует иметь в виду у детей с эмбриональной или пупочной грыжей, макроглосией и эмбриональными опухолями (нейробластома, опухоль Вильмса).
Возможные осложнения у больных с синдромом Беквита-Видеманна:
- вероятность неонатальной гипогликемии (60%) с развитием судорог в послеоперационном периоде;
- большой процент (10-40%) эмбриональных опухолей, особенно при нефромегалии или соматической асимметрии туловища, которое нуждается в наблюдении и проведении ультразвукового исследования почек 3 раза в год до 3- летнего возраста и у старших детей 2 раза в год до 14 лет (своевременная диагностика опухоли Вильмса).
Синдром Марфана — доминантный синдром. Минимальные диагностические критерии — астеническое телосложение, доли-хостеномиелия (длинные конечности), арахнодактилия, подвывих хрусталика или слабость ресничного пояска (цинновой связки) и отсутствие гомоцистина в моче. Диагноз синдрома Марфана следует иметь в виду у детей с врожденными деформациями грудной клетки, аномалиями позвоночника (сколиоз, кифоз), патологической подвижностью суставов и различными грыжами ( паховая, пупочная, диафрагмальная). Очень часто при этом заболевании встречается специфический признак патологии соединительной ткани — пролапс митрального клапана.
Особенности ведения и осложнения у больных с синдромом Марфана:
• затруднение интубации из-за подвижности височно-нижнечелюстного сустава и суставов шейного отдела позвоночника;
• опасность внезапного повышения или снижения артериального давления во время операции;
• осторожное применение мышечных релаксантов при миопатических проявлениях (возможен парадоксальный или пролонгированный эффект);
• возможность летальной желудочковой аритмии и бактериального эндокардита в послеопераиионном периоде при пролапсе митрального клапана;
• расширение дуги аорты, образование аневризм и расслоения аорты с возможностью разрыва;
• повышенный риск спонтанного пневмоторакса (4,4%);
• повышенная частота пневмоний и хронических эмфиземоподобных изменений;
• снижение жизненной емкости легких, увеличивающее риск анестезиологических осложнений.
Примерно у 1% новорожденных имеется неслучайное сочетание нескольких малых аномалий развития и врожденных дефектов, из которых в 40% можно диагностировать тот или иной синдром, а в 60% случаев требуется выделение так называемых новых синдромов.
Большую роль в решении проблем диагностики врожденных аномалий играет антенатальное обследование, которое проводится с 14-18 недели беременности. Оно разрешает виявить основные виды пороков развития задолго до рождения ребенка. В случае антенатальной диагностики пороков будущие родители должны быть подробно, доступным языком проинформированы о заболевании будущего ребенка и возможности коррекции этого порока развития. На основании этой информации они имеют право решить судьбу своего еще не родившегося малыша.
Сроки возможной эхографической визуализации некоторых ВВР
Вид ВВР |
Срок в неделях |
Доступ |
Автор |
Акрания |
11 |
PV |
Kennedy K.A. et all |
Цефалоцеле |
13 |
PV |
Bronstein m, Zinner M.Z. |
Вентрикуломегалия |
После 14 |
Абд. |
Мєдвєдєв М.В. |
Аномалия Dandy-Walker |
13 |
PV |
Achiron R., Achiron A. |
Галопрозенцефалия |
12 |
PV |
Воронін Д.В. |
Spina вifida |
12 |
PV |
Sebire N. et al. |
Расщелина губы и неба |
12 |
PV |
Воронін Д.В. |
Диафрагмальная грижа |
14 |
PV |
Bocain M. et al. |
ВПС |
11 |
PV |
Gembruch U. et al. |
Эктопия сердца |
10 |
PV |
Воронін Д.В. |
Атрезия пищевода |
С 26 нед. |
Абд. |
Мєдвєдєв М.В. |
Дуоденальная атрезия |
14 |
PV |
Petrikovsky B.M. |
Атрезия тонкого кишечника |
С 26 нед. |
Абд. |
Мєдвєдєв М.В |
Атрезия ануса |
После 30 |
Абд. |
Harris B. et al. |
Изолированный асцит |
14 |
Абд. |
Мєдвєдєв М.В. |
Гастрошизис |
10 |
PV |
Воронін Д.В. |
Омфалоцеле |
После 13 |
PV |
Van Zalen-Sprock R.M. et al. |
Гидронефроз |
13 |
PV |
Nicolaides K.N. |
Мегалоцистис |
10 |
PV |
Sebire N. et al. |
Агенезия почек |
13 |
PV |
Bronstein M., Zinner E.Z. |
Поликистоз почек |
19 - 20 |
Абд. |
Wisser J. et al. |
Ахондроплазия |
24 - 28 |
Абд. |
Мєдвєдєв М.В., Юдіна О.В. |
Полидактилия |
14 |
PV |
Bronstein M., Zinner E.Z. |
Крестцово-копчиковая тератома |
13 |
PV |
Мєдвєдєв М.В. |
Симптомы при ультразвуковом обследовании которые определяются у плода.
Полигидроамнион (желудочно-кишечная обструкция, дефекты брюшной стенки, аненцефалия, диафрагмальная грыжа, неспособность плода глотать и концентрировать мочу).
Дистоция (непроходимость кишечника, дефект брюшной стенки, аномалии мочевыводящей системы).
Мекониевый перитонит (обструкция кишечника, перфорация кишечника).
Асцит плода (аномалии мочевыдительной системы, перитонит, сердечные заболевания).
Олигогидроамнион (агенезия почек).
Расширение участков кишечной трубки.
Отсутствие визуализации желудка и участков кишечника при повторных обследованиях.
Патология, при которой возможная внутриутробная хирургическая коррекция:
Диафрагмальная грыжа небольших размеров
Гидроцефалия
Гигрома шеи
Гидроторакс
Омфалоцеле
Гидронефроз
Мегауретер
Атрезия уретры
Стеноз аорты и легочной артерии
Патология, которая требует рождения путем Кесаревого сечения: гигантская тератома, омфалоцеле, гастрошизис, лимфангиома шеи больших размеров.
Возможна и обязательна антенатальная диагностика пороков, так как около 45% больных имеют объединенные пороки развития, которые нередко являются ведущими в танатогенезе.
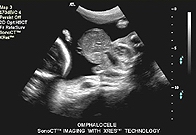 Рис.
1. Сонограмма плода с омфалоцеле
Рис.
1. Сонограмма плода с омфалоцеле
Возможна антенатальная диагностика гастрошизиса. Этот порок редко объединяется с другими пороками развития.
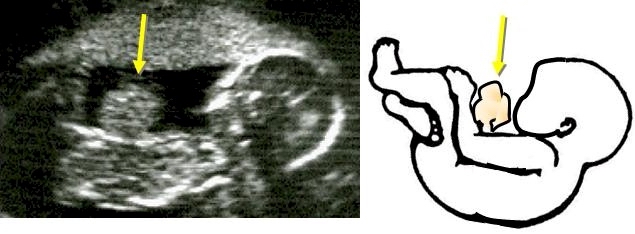
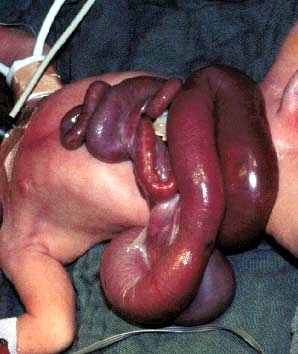
Рис. 2. Сонограмма плода с гастрошизисом, новорожденный с гастрошизисом.
Возможна и обязательна антенатальная диагностика врожденных пороков, так как у около 50% больных с синдром Дауна, например, определяется сочетание с высокой кишечной непроходимостью (ВКН). Причины ВКН: атрезия двенадцатиперстной кишки, кольцевидная поджелудочная железа, мембрана двенадцатиперстной кишки (часто объединяется с мальротацией). Редчайшие формы пороков - это предуоденальна портальная вена, аберрантный сосуд печени, удвоение двенадцатиперстной кишки.
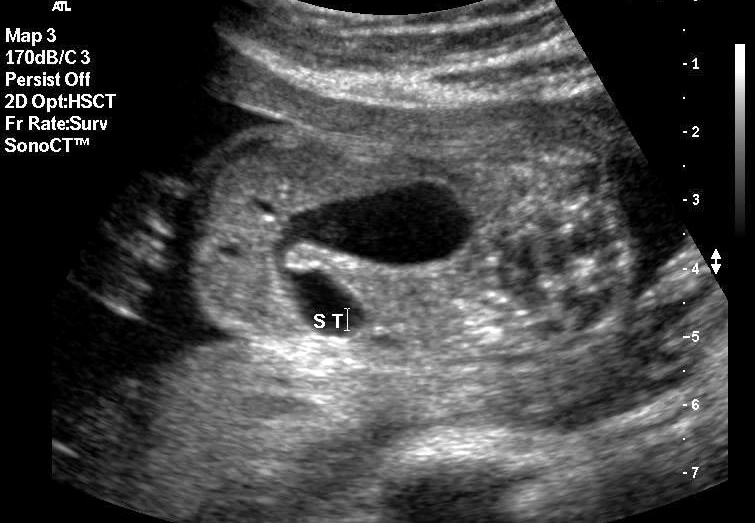

Рис. 3. Сонограмма плода и рентгенограмма ребенка с ВКН.
Патология, которая дает основания к обсуждению вопроса о прерывании беременности: ахондроплазия, клапан задней уретры с двусторонним мегауретером, гидронефрозом, кистозная дисплазия почек, грубые инвалидизурующие аномалии конечностей, любые аномалии ЦНС до 20-22 недель (за исключением кист сосудистых сплетений), множественные пороки развития.
Патология, которая требует прерывания беременности: аненцефалия, голопрозенцефалия, экзенцефалия, гидроцефалия, обусловленная синдромом Арнольда-Киари, и спинномозговые грыжи больших размеров, внутричерепные опухоли, поренцефалия больше 1см, массивные ВЧК, расщепление лица, агенезия глазных яблок, грубые пороки сердца: эктопия сердца, двойное отхождение сосудов, ОАС, выраженная гипоплазия желудочков сердца, общий жнлудочек, агенезия почек, поликистоз детского типа, агенезия желчных протоков, сращенная двойня.
Патология, которая требует неотложного после рождения ребенка перевода в хирургическое отделение: гастрошизис, омфалоцеле (эмбриональные грыжи с узким основанием), атрезия пищевода, атрезия тонкого и толстого кишечника, неперфорированный анус, диафрагмальная грыжа, кистозная гипоплазия легкого с ДН, выраженное сужение уретры, опухолеобразные образованиея, которые приводят к асфиктическому синдрому.
Патология, которая требует госпитализации в хирургическое отделение в периоде новонароджености: объемные образования брюшной полости, легочная секвестрация, мультикистоз почки, мегауретер, гидронефроз, экстрофия мочевого пузыря, тератомы крестцово-копчиковые, кисты или атрезия желчных путей.
Противопоказание к оперативному вмешательству у детей с пороками развития.
Абсолютно противопоказаны операции детям, у которых из-за имеющегося у них порока, они являются нежизнеспособным. Не следует начинать операцию у ребенка, который находится в преагональном и агональном состоянии или в состоянии шока ІІІ и ІV степени, но после вывода его из этого состояния при наличии абсолютных показаний оперативное вмешательство возможно.
В тех случаях, когда причина тяжелого состояния может быть устранена только хирургическим путем (кровотечение, пневмоторакс и т.п. ), операцию можно начинать и до окончательного вывода пациента из шока на фоне противошоковых мероприятий.
Кашель и насморк у ребенка, которые носят хронический характер и не сопровождаются повышением температуры и отсутствием аппетита, не являются противопоказанием к оперативному вмешательству, в том случае, когда катаральные явления возникают остро и сопровождаются температурной реакцией и другими признаками какого-нибудь заболевания, операцию следует отложить.
Относительными противопоказаниями считают заболевания дыхательных путей, инфекционные болезни, нарушение нормального развития ребенка, связанные с недостаточным питанием, поносом и другими причинами: эксудативный диатез, пиодермия, резко выраженные явления рахита, состояние после вакцинации, повышение температуры неясной этиологии.
Развитие анестезиологии и реаниматологии расширило возможности хирургических вмешательств даже при очень трудном состоянии больных. Кроме того, некоторые дети на протяжении многих месяцев страдают каким-нибудь заболеванием дыхательных путей, а продолжительное откладывание операции опасно, или оказывает содействие респираторным заболеваниям (например, при незаращении неба). В таких случаях проводят вмешательство, как только появился светлый промежуток и стихли катаральные явления.
Оперируют детей лишь по согласию родителей или людей, которые их заменяют. Письменное согласие фиксируют в истории болезни. В крайнем случае можно обойтись устным согласием, данным при свидетелях. Если есть абсолютные показания к операции, а родителям не удается сообщить об этом и их согласие не получено, вопрос об операции решается консилиумом с 2-3мя врачами, о чем извещают главного врача.
МАТЕРИАЛЫ ДЛЯ САМОКОНТРОЛЯ
Ситуационные задачи.
Задача 1. Соответственно международной классификации, все врожденные пороки развития подразделяются на 4 группы.
1. Какие группы врожденных пороков выделяются согласно международной классификации?
2. Определите понятие врожденный порок.
3. Определите понятие дизрупция.
4. Определите понятие деформации.
5. Определите понятие дисплазии.
Задача 2. Малая аномалия развития - редчайший вариант строения тела или врожденная аномалия, которая не имеет медицинского значения, не нуждается в лечении. Для детского хирурга важную роль играет определение порока развития или аномалии развития для уточнения тактики лечения и разработки реабилитационных мероприятий
1. Определите понятие малые пороки развития.
2. Назовите основные группы малых аномалий у детей.
3. Обоснуйте определение врожденного порока.
4. Обоснуйте определение синдрома.
5. Реабилитация детей с малыми пороками развития.
Задача 3. У ребенка выявлен синдром, который основывается на объединении низкого роста, широкой и крылообразной шеи, деформации грудной клетки, врожденного порока сердца и необычного лицевого фенотипа: лобные бугры, гипертелоризм, ротированные ушные раковины со свисающими завитками.
1. Определение синдрома этого синдрома.
2. Пороки развития, которые определяются у детей с этим синдромом.
3. Практические рекомендации для хирургов.
4. Прогноз для детей с данным пороком.
5. Реабилитация детей с данным синдромом.
Задача 4. У мальчика астеническое телосложение, долихостеномиелия (длинные конечности), арахнодактилия, подвывих хрусталика и отсутствие гомоцистина в моче. Во время осмотра определяется воронкообразная деформация грудной клетки, кифосколиоз, патологическая подвижность суставов.
1. Определить понятие синдром.
2. Какие деформации характерны для этого синдрома?
3. Особенности ведения детей с этим синдромом.
4. Какие осложнения можно наблюдать у детей с этим синдромом?
5. Реабилитация детей с этим синдромом.
Задача 5. У новорожденных с пороками развития выделяются разные группы, которые различаются в подходах к тактике ведения и лечения.
1. Сформулируйте основные показания при пороках развития, которые дают основания к обсуждению вопроса о прерывании беременности.
2. При каких пороках развития показано прерывание беременности?
3. При каких пороках новорожденного следует немедленно перевести в хирургическое отделение?
4. При каких пороках новорожденного следует перевести в хирургическое отделение в период новорожденности?
Задача 6. У ребенка наследственное аутосомно-доминантное заболевание, основанное на объединении низкого роста, крылообразной шеи, деформации грудной клетки, врожденного порока сердца и необычного лицевого фенотипа – лобные бугры, гипертелоризм, ротированные ушные раковины со свисающими завитками, крипторхизм. О каком синдроме следует думать прежде всего.
1. Определите основные проявления этого синдрома.
2. Этиология, патогенез заболевания.
3. Лечение детей с этим синдромом.
4. Назовите осложнения, которые могут возникнуть у ребенка во время оперативного лечения.
5. Реабилитация детей с этим синдромом.
Задача 7. У беременной на 24 неделе при обследовании выявили у плода эмбриональную грыжу больших размеров, других пороков развития у ребенка нет.
1. Этиология, патогенез заболевания.
2. Принципы сбора генетического анамнеза.
3. Тактика ведения беременной и прием родов.
4. Первая медицинская и неотложная помощь новорожденному с эмбриональной грыжей.
5. Лечебная тактика у детей с врожденными пороками развития.
Задача 8. У новорожденной девочки выявлены малые аномалии развития: уплощенный затылок, легкий птоз, примитивная форма ушных раковин, аномальная форма завитка уха, полидактилия кисти.
1. Определите понятие о малой аномалии развития.
2. Классификация малых аномалий развития.
3. Тактика обследования новорожденного с малыми пороками развития.
4. Тактика ведения и лечения ребенка с малыми пороками развития.
5. Реабилитация детей с пороками развития.
Задача 9. У новорожденного ассоциация пороков развития, при которой выявлен порок развития позвоночника, атрезия ануса, трахео-эзофагиальный свищ.
1. Определите синдром при котором наблюдается эта ассоциация.
2. Основные проявления этого синдрома.
3. Диагностика синдрома.
4. Неотложная помощь и лечебная тактика синдрома.
5. Реабилитация детей.
Задача 10. У новорожденного на 5 сутки при клиническом обследовании выявили тромбоцитопению. Кроме этого у ребенка радиальная аплазия с сохраненным первым пальцем и порок сердца.
1. Определите понятие о синдроме
2. Диагностическая тактика у детей с этим синдромом.
3. Неотложная помощь и лечебная тактика у детей с этим синдромом
4. Осложнения, которые могут возникнуть у ребенка с этим синдромом.
Тестовые задачи
1. Патология, которая дает основания к обсуждению вопроса о прерывании беременности по данным ультразвукового исследования в 18-20 недель беременности, это:
А. Врожденный вывих бедра
В. Грубые инвалидизирующие аномалии конечностей
С. Синдактилия
D. Полидактилия
Е. Гипоспадия
2. Патология, которая требует неотложного перевода новорожденного в хирургическое отделение при пороке развития, которую диагностировали перед родами при ультразвуковом исследовании.
А. Гастрошизис
В. Асимметрия лица
С. Деформация позвоночника
D. Полидактилия
Е. Отсутствие одной почки
3. Абсолютно противопоказаны операции детям, которые из-за имеющихся у них пороков являются нежизнеспособными.
A. Асфиксия
B. Новорожденный в состоянии комы ІІ
C. Гипотрофия
D. Новорожденный в состоянии комы ІІІ - ІV степени
E. Новорожденный в состоянии комы І степени
4. У новорожденного определен порок развития конечности, малая аномалия сердца, лица. Неонатолог определил это состояние как синдром. Какое основное определение синдрома.
A. Врожденный порок развития
B. Малая аномалия развития
C. Аномалия развития
D. Деформация
E. Наличие у ребенка двух симптомов и больше
5. Синдром Дауна - это симптомокомплекс у новорожденного при котором определяются следующие симптомы:
A. Мышечный гипертонус, микроглосия, обычное лицо, монголоидный разрез глаз, отсутствие V пальцев
B. Мышечная гипотония, макроглосия, луноподобое лицо, монголоидный разрез глаз, гипоплазия V пальцев
C. Мышечная гипотония, обычное лицо лица, обычный разрез глаз, гипоплазия І пальца
D. Мышечный тонус обычный, микроглосия, деформация лица, обычный разрез глаз
E. Мышечный гипертонус, синдактилия, деформация лица, птоз обеих глаз, макроглосия
6. У ребенка отмечена большая масса при рождении и постнатальное опережение физического развития, дефекты закрытия передней стенки живота - пупочная грыжа, диастаз прямых мышц живота, нефромегалия, гепатомегалия, спленомегалия, макроглосия, гемангиома кожи лба, "насечки" на ушных раковинах. Какой синдром у ребенка?
A. Синдром Марфана
B. Синдром Беквита-Видеманна
C. Синдром Дауна
D. Синдром Элерса-Данлоса
E. Синдром ТАР
7. У ребенка наследственное аутосомно-доминантое заболевание, основанное на объединении низкого роста, крылообразной шеи, деформации грудной клетки, врожденного порока сердца и необычного лицевого фенотипа – лобные бугры, гипертелоризм, роторованные ушные раковины со свисающими завитками, крипторхизм. О каком синдроме следует думать прежде всего.
A. Синдром Беквита-Видеманна
B. Синдром ТАР
C. Синдром Элерса-Данлоса
D. Синдром Дауна
E. Синдром Нунан
8. У новорожденного есть такие клинические проявления заболевания - тромбоцитопения, радиальная аплазия. Определено аутосомно-рецессивне наследование заболевания, которое характеризуется соединением тромбоцитопении с двусторонним отсутствием лучевой кости с сохранением первого пальца кисти, наличием врожденного порока сердца. О каком синдроме у ребенка следует думать прежде всего.
A. Синдром Беквита-Видеманна
B. Синдром ТАР
C. Синдром Дауна
D. Синдром Нунан
E. Синдром Элерса-Данлоса
9. В новорожденного - аномальная форма правой нижней конечности и асимметрия лица, вызванные механическим давлением в период внутриутробного развития. У матери диагностирована фибромиома матки до беременности. Этот порок следует определить как проявление:
A. Дисплазии
B. Дизрупции
C. Деформация
D. Остеодисплазии
E. Аномалии
10. У ребенка трех лет во время обследования выявили недоразвитие левой почки, которое проявляется уменьшением размеров, превышающим отклонение от средних показателей для данного возраста. Эту аномалию у ребенка следует определить как:
A. Дисплазия почки
B. Аплазия почки
C. Врожденная гипоплазия
D. Нефроптоз слева
E. Врожденная гипертрофия
Перечень теоретических вопросов
1. Пороки развития у детей, этиология, патогенез, частота.
2. Синдромология в детской хирургии.
3. Малые аномалии развития.
4. Синдром Дауна, синдром Эдвардса,VATER ассоциация, синдром Опиця, синдром, CHARGE.
5. Особенности оперативных вмешательств у детей с синдромами и пороками развития.
6. Профилактика пороков развития.
Практические задания:
1. Методика обследования ребенка и определение малых пороков развития.
2. Анализировать УЗИ при пороках развития у детей.
Рекомендованная литература
Основная литература:
1. Сушко В.І. Хірургія дитячого віку. Київ. “Здоров’я”, 2009, 65-70, 325- 379.
2. Амбулаторно-поліклінічна допомога дітям. Під редакцією проф. Сушка В.І. 2004,
3. Ленюшкин А.И. Руководство по детской поликлинической хирургии. - Л.: Медицина, 1986. - 108 с
Дополнительная литература:
1. Исаков Ю.Ф. Хирургические болезни у детей. - М.:Медицина, 1998.-248 с.
2. Ашкрафт К.У., Холдер Т.М. Детская хирургия /Пер. с англ. - СПб., Хардфорд, 1996.– 458с.
3. Andrews NC. Disorders of Iron Metabolis. The New England Journal of Medicine, 341(26):1986-1995, 1999
4. Cohen M.M.,Jr: The Child With Multiple Birth Defects. New York: Raven Press. 1982. 189 pp.
5. Cohen M.M., Jr: The Child With Multiple Birth Defects. Second edition. New York: Oxford University Press. 1997. 267 pp.
6. Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative hemochromatosis mutations. J Med Genet, 34:275-278, 1997
7. Olynyk JL, Cullen DJ, Aquilia S, Rossi E, Summerville L, and Powell LW. A Population-Based Study of the Clinical Expression of the Hemochromatosis Gene. The New England Journal of Medicine, 341(10):718-724, 1999
8. Opitz J.M.: Terminological and epistemological considerations of human malformations. In Harris H. and Hirschhorn K.(eds): Advances in Human Genetics. New York:Plenum, 1979, pp.71-107
9. Spranger J., Benirschke K., Hall J.G., et al.: Errors of morphogenesis: Concepts and terms. Recommendations of an International Working Group // J.Pediatr. 100(1):160-165, 1982
10. Stevenson R.E., Hall J.G., Goodman R.M. Human Malformation and Related Anomalies.Vol. I and Vol.II. New York & Oxford: Oxford University Press, 1993, vol. I - 271 p.; vol. II - 1162 p.
12. Делоне Н.Л., Солониченко В.Г. Адаптивные фенотипы человека в физиологии и медицине. Успехи физиологических наук, 1999, т.30, № 2, с.50-62.
13. Солониченко В.Г., Красовская Т.В. Дисморфология хирургических болезней у детей. Детская хирургия, 1998, № 4, с.4-9.