

64 Ресинтез
Р![]() есинтез
липидов в энтероцитах.
После всасывания продуктов гидролиза
жиров жирные кислоты и 2-моноацилглицеролы
в клетках слизистой оболочки тонкого
кишечника включаются в процесс ресинтеза
с образованием триацилглицеролов.
Жирные кислоты вступают в реакцию
этерификации только в активной форме
в виде производных коэнзима А, поэтому
первая стадия ресинтеза жиров - реакция
активации жирной кислоты:
есинтез
липидов в энтероцитах.
После всасывания продуктов гидролиза
жиров жирные кислоты и 2-моноацилглицеролы
в клетках слизистой оболочки тонкого
кишечника включаются в процесс ресинтеза
с образованием триацилглицеролов.
Жирные кислоты вступают в реакцию
этерификации только в активной форме
в виде производных коэнзима А, поэтому
первая стадия ресинтеза жиров - реакция
активации жирной кислоты:
HS КоА + RCOOH + АТФ → R-CO ~ КоА + АМФ + Н4Р2О7.
Реакция катализируется ферментом ацил-КоА-синтетазой (тиокиназой). Затем ацил~КоА участвует в реакции этерификации 2-моноацилглицерола с образованием сначала диацилгли-церола, а затем триацилглицерола. Реакции ресинтеза жиров катализируют ацилтранеферазы.Ресинтез липидов – это синтез липидов в стенке кишечника из поступающих сюда экзогенных жиров, иногда могут использоваться и эндогенные жирные кислоты. Основная задача этого процесса – связать поступившие с пищей средне- и длинноцепочечные жирные кислоты со спиртом – глицеролом или холестеролом. Это ликвидирует их детергентное действие на мембраны и позволит переносить по крови в ткани. Поступившая в энтероцит жирная кислота обязательно активируется через присоединение коэнзима А. Образовавшийся ацил-SКоА участвует в реакциях синтеза эфиров холестерола, триацилглицеролов и фосфолипидов.
Р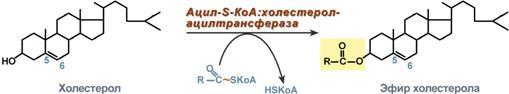 есинтез
эфиров холестерола.
Холестерол этерифицируется с использованием
ацил-S-КоА и фермента
ацил-КоА:холестерол-ацилтрансферазы
(АХАТ). Реэтерификация холестерола
напрямую влияет на его всасывание в
кровь. В настоящее время ищутся возможности
подавления этой реакции для снижения
концентрации ХС в крови.
есинтез
эфиров холестерола.
Холестерол этерифицируется с использованием
ацил-S-КоА и фермента
ацил-КоА:холестерол-ацилтрансферазы
(АХАТ). Реэтерификация холестерола
напрямую влияет на его всасывание в
кровь. В настоящее время ищутся возможности
подавления этой реакции для снижения
концентрации ХС в крови.
Р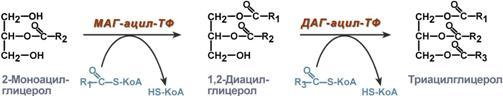 есинтез
триацилглицеролов.
Для ресинтеза ТАГ есть два пути: Первый
путь,
основной – 2-моноацилглицеридный
– происходит при участии экзогенных
2-МАГ и ЖК в гладком эндоплазматическом
ретикулуме энтероцитов: мультиферментный
комплекс триацилглицерол-синтазы
формирует ТАГ. Поскольку 1/4 часть ТАГ в
кишечнике полностью гидролизуется и
глицерол в энтероцитах не задерживается,
то возникает относительный избыток
жирных кислот для которых не хватает
глицерола. Поэтому существует второй,
глицеролфосфатный, путь в шероховатом
эндоплазматическом ретикулуме. Источником
глицерол-3-фосфата служит окисление
глюкозы, так как пищевой глицерол быстро
покидает энтероциты и уходит в кровь.
Здесь можно выделить следующие реакции:
1. Образование глицерол-3-фосфата из
глюкозы. 2.Превращение глицерол-3-фосфата
в фосфатидную кислоту. 3.Превращение
фосфатидной кислоты в 1,2-ДАГ. 3.Синтез
ТАГ.
есинтез
триацилглицеролов.
Для ресинтеза ТАГ есть два пути: Первый
путь,
основной – 2-моноацилглицеридный
– происходит при участии экзогенных
2-МАГ и ЖК в гладком эндоплазматическом
ретикулуме энтероцитов: мультиферментный
комплекс триацилглицерол-синтазы
формирует ТАГ. Поскольку 1/4 часть ТАГ в
кишечнике полностью гидролизуется и
глицерол в энтероцитах не задерживается,
то возникает относительный избыток
жирных кислот для которых не хватает
глицерола. Поэтому существует второй,
глицеролфосфатный, путь в шероховатом
эндоплазматическом ретикулуме. Источником
глицерол-3-фосфата служит окисление
глюкозы, так как пищевой глицерол быстро
покидает энтероциты и уходит в кровь.
Здесь можно выделить следующие реакции:
1. Образование глицерол-3-фосфата из
глюкозы. 2.Превращение глицерол-3-фосфата
в фосфатидную кислоту. 3.Превращение
фосфатидной кислоты в 1,2-ДАГ. 3.Синтез
ТАГ.
Р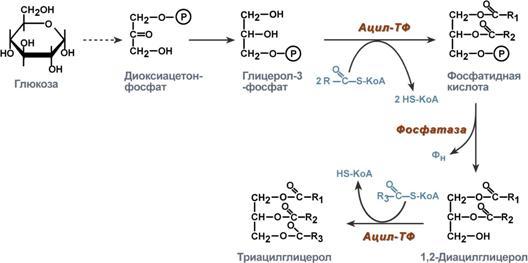 есинтез
фосфолипидов.
Фосфолипиды синтезируются также как и
в остальных клетках организма. Для этого
есть два способа. Первый – с использованием
1,2-ДАГ и активных форм холина и этаноламина
для синтеза фосфатидилхолина или
фосфатидилэтаноламина. Второй путь –
через синтезируемую in situ фосфатидную
кислоту. После ресинтеза фосфолипиды,
триацилглицеролы, холестерол и его
эфиры упаковываются в особые транспортные
формы липидов – липопротеины и только
в такой форме они способны покинуть
энтероцит. В кишечнике формируются два
вида липопротеинов – хиломикроны и
липопротеины высокой плотности.
есинтез
фосфолипидов.
Фосфолипиды синтезируются также как и
в остальных клетках организма. Для этого
есть два способа. Первый – с использованием
1,2-ДАГ и активных форм холина и этаноламина
для синтеза фосфатидилхолина или
фосфатидилэтаноламина. Второй путь –
через синтезируемую in situ фосфатидную
кислоту. После ресинтеза фосфолипиды,
триацилглицеролы, холестерол и его
эфиры упаковываются в особые транспортные
формы липидов – липопротеины и только
в такой форме они способны покинуть
энтероцит. В кишечнике формируются два
вида липопротеинов – хиломикроны и
липопротеины высокой плотности.
П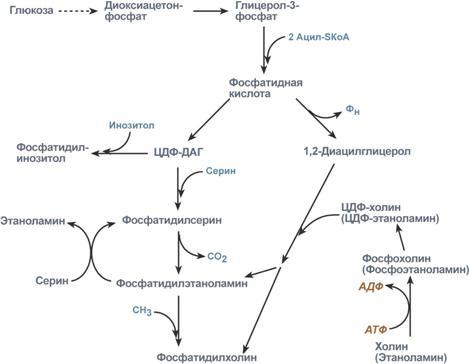 оскольку
липиды являются в основе своей гидрофобными
молекулами, то они транспортируются в
водной фазе крови в составе особых
частиц – липопротеинов. Такие транспортные
липопротеины можно сравнить с орехом,
который имеет скорлупу и ядро. Поверхность
липопротеиновой частицы ("скорлупа")
гидрофильна и сформирована белками,
фосфолипидами и свободным холестеролом.
Триацилглицеролы и эфиры холестерола
составляют гидрофобное ядро. Липопротеины
различаются по соотношению триацилглицеролов,
холестерола и его эфиров, фосфолипидов
и как сложные белки состоят из четырех
классов.1. хиломикроны (ХМ), 2.липопротеины
очень низкой плотности (ЛПОНП,
пре-β-липопротеины, пре-β-ЛП), 3. липопротеины
низкой плотности (ЛПНП, β-липопротеины,
β-ЛП), 4. липопротеины высокой плотности
(ЛПВП, α-липопротеины, α-ЛП). Транспорт
триацилглицеролов от кишечника к тканям
(экзогенные ТАГ) осуществляется в виде
хиломикронов
(ХМ),
от печени к тканям (эндогенные ТАГ) – в
виде липопротеинов очень низкой
плотности. В транспорте ТАГ к тканям
можно выделить последовательность
следующих событий:
оскольку
липиды являются в основе своей гидрофобными
молекулами, то они транспортируются в
водной фазе крови в составе особых
частиц – липопротеинов. Такие транспортные
липопротеины можно сравнить с орехом,
который имеет скорлупу и ядро. Поверхность
липопротеиновой частицы ("скорлупа")
гидрофильна и сформирована белками,
фосфолипидами и свободным холестеролом.
Триацилглицеролы и эфиры холестерола
составляют гидрофобное ядро. Липопротеины
различаются по соотношению триацилглицеролов,
холестерола и его эфиров, фосфолипидов
и как сложные белки состоят из четырех
классов.1. хиломикроны (ХМ), 2.липопротеины
очень низкой плотности (ЛПОНП,
пре-β-липопротеины, пре-β-ЛП), 3. липопротеины
низкой плотности (ЛПНП, β-липопротеины,
β-ЛП), 4. липопротеины высокой плотности
(ЛПВП, α-липопротеины, α-ЛП). Транспорт
триацилглицеролов от кишечника к тканям
(экзогенные ТАГ) осуществляется в виде
хиломикронов
(ХМ),
от печени к тканям (эндогенные ТАГ) – в
виде липопротеинов очень низкой
плотности. В транспорте ТАГ к тканям
можно выделить последовательность
следующих событий:
1)Образование незрелых первичных ХМ в кишечнике.
2)Движение первичных ХМ через лимфатические протоки в кровь.
3)Созревание ХМ в плазме крови – получение белков апоС-II и апоЕ от ЛПВП.
4)Взаимодействие с липопротеинлипазой (ЛПЛ) эндотелия, которая отщепляет жирные кислоты от ТАГ. Жирные кислоты переходят непосредственно в данную ткань или, связываясь с альбумином, разносятся по организму. В результате количество ТАГ в хиломикроне резко снижается и образуются остаточные ХМ.
5)Переход остаточных ХМ в гепатоциты и полный распад их структуры.
6)Синтез ТАГ в печени из пищевой глюкозы. Использование ТАГ, пришедших в составе остаточных ХМ.
7)Образование первичных ЛПОНП в печени.
8)Созревание ЛПОНП в плазме крови – получение белков апоС-II и апоЕ от ЛПВП.
9)Взаимодействие с липопротеинлипазой эндотелия и потеря большей части ТАГ. Образование остаточных ЛПОНП (по-другому липопротеины промежуточной плотности, ЛППП).
10)Остаточные ЛПОНП переходят в гепатоциты и полностью распадаются, либо остаются в плазме крови. После воздействия на них печеночной ТАГ-липазы в синусоидах печени ЛПОНП превращаются в ЛПНП.
Характеристика хиломикронов. Общая характеристика: -формируются в кишечнике из ресинтезированных жиров, -в их составе преобладают ТАГ, мало белка, фосфолипидов и холестерола (2% белка, 87% ТАГ, 2% ХС, 5% эфиров ХС, 4% фосфолипидов), - основным апобелком является апоВ-48, это структурный липопротеин, в плазме крови получают от ЛПВП белки апоС-II и апоЕ, - в норме натощак не обнаруживаются, в крови появляются после приема пищи, поступая из лимфы через грудной лимфатический проток, и полностью исчезают через 10-12 часов,- не атерогенны.
Функция: Транспорт экзогенных ТАГ из кишечника в ткани, запасающие или использующие жиры, в основном жировую ткань, легкие, печень, миокард, лактирующую молочную железу, костный мозг, почки, селезенку, макрофаги. На эндотелии капилляров этих тканей имеется фермент липопротеинлипаза.
М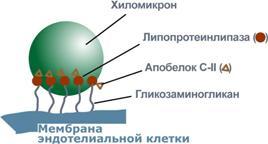 етаболизм
1. После ресинтеза жиров в эпителиоцитах
кишечника формируются первичные
хиломикроны, имеющие только апоВ-48.
етаболизм
1. После ресинтеза жиров в эпителиоцитах
кишечника формируются первичные
хиломикроны, имеющие только апоВ-48.
2. Из-за большого размера они не проникают напрямую в кровеносное русло и эвакуируются через лимфатическую систему, попадая в кровь через грудной лимфатический проток.
3. В крови хиломикроны взаимодействуют с ЛПВП и приобретают от них апоС-II и апоЕ, образуя зрелые формы. Белок апоС-II является активатором фермента липопротеинлипазы, белок апоЕ необходим для удаления из крови остаточных хиломикронов.
4. На эндотелии капилляров вышеперечисленных тканей находится фермент липопротеинлипаза (ЛПЛ), отщепляющий жирные кислоты от ТАГ. Количество фермента увеличивается при действии инсулина и прогестерона.
5. После взаимодействия хиломикрона с ферментом триацилглицеролы, находящиеся в составе хиломикронов, гидролизуются с образованием свободных жирных кислот. Жирные кислоты перемещаются в клетки органа, либо остаются в плазме крови и в комплексе с альбумином разносятся с кровью в другие ткани. Липопротеинлипаза способна удалить до 90% всех ТАГ, находящихся в хиломикроне или ЛПОНП.
6. После окончания работы ЛПЛ остаточные хиломикроны попадают в гепатоциты посредством апоЕ-рецепторного эндоцитоза и разрушаются.
Содержание липидов в кровотоке может понижаться вслед¬ствие откладывания их в различных тканях. Способность откладывать жир характерна для всех тканей, кроме мозга. Главную роль в обмене липидов играют жировая ткань и печень. Количество жиро¬вой ткани нарастает с возрастом. Пределы конц жиров в крови:Норма-4-8г/л, если больше-гиперлипемия( при сах диабете, ожирении). В норме концентрация жиров в крови колеблется в довольно широких пределах — 10-200 мг/дл, в среднем около 0,1 %. Отметим для сравнения, что концентрация жиров в молоке равна примерно 3 %. После приема пищи концентрация хиломикронов в крови повышается, достигает максимума примерно через 5 ч, затем начинает снижаться. Сходным образом, но с меньшей амплитудой изменяется концентрация ЛОНП в крови. При этом надо отметить, что концентрация хиломикронов в большей мере зависит от содержания жиров в пище, а концентрация ЛОНП — от содержания углеводов. В крови, взятой для анализа утром до завтрака, т. е. после большого ночного перерыва в приеме пищи, хиломикроны не обнаруживаются, а концентрация ЛОНП минимальна. Пропорционально концентрации этих липопротеинов снижена и концентрация жиров в крови.При обычном ритме питания и небольших физических нагрузках в крови в дневное время постоянно имеются хиломикроны и ЛОНП, поскольку время переваривания жиров мало отличается от времени между приемами пищи.
65 Использование экзогенных жиров тканями
В. Использование экзогенных жиров тканями
Действие липопротеинлипазы на ХМ. В крови триацилглицеролы, входящие в состав зрелых ХМ, гидролизуются ферментом липопротеин-липазой, или ЛП-липазой (рис. 8-20). ЛП-липа-за связана с гепарансульфатом (гетерополисаха-ридом), находящимся на поверхности эндотелиальных клеток, выстилающих стенки капилляров кровеносных сосудов. ЛП-липаза гидролизует молекулы жиров до глицерола и 3 молекул жирных кислот. На поверхности ХМ различают 2 фактора, необходимых для активности ЛП-липазы - апоС-П и фосфолипиды. АпоС-П активирует этот фермент, а фосфолипиды участвуют в, связывании фермента с поверхностью ХМ.
ЛП-липаза синтезируется в клетках многих тканей: жировой, мышечной, в лёгких, селезёнке, клетках лактирующей молочной железы. Изоферменты ЛП-липазы в разных тканях отличаются по значению Кm: ЛП-липаза жировой ткани имеет в 10 раз более высокое значение Кm, чем, например, ЛП-липаза сердца, поэтому гидролиз жиров ХМ в жировой ткани происходит в абсорбтивный период. Жирные кислоты поступают в адипоциты и используются для синтеза жиров. В постабсорбтивном состоянии, когда количество жиров в крови снижается, ЛП-липаза сердечной мышцы продолжает гидролизовать жиры в составе ЛПОНП, которые присутствуют в крови в небольшом количестве, и жирные кислоты используются этой тканью как источники энергии, даже при низкой концентрации жиров в крови. ЛП-липазы нет в печени, но на поверхности клеток этого органа имеется другой фермент - печёночная липаза, не действующая на зрелые ХМ, но гидролизующая жиры в ЛППП, которые образуются из ЛПОНП.
Судьба жирных кислот, глицерола и остаточных хиломикронов. В результате действия ЛП-липазы на жиры ХМ образуются жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткани (рис. 8-20). В жировой ткани в абсорбтивный период жирные кислоты депонируются в виде триацилглицеролов, в сердечной мышце и работающих скелетных мышцах используются как источник энергии. Другой продукт гидролиза жиров, глицерол, растворим в крови, транспортируется в печень, где в абсорбтивный период может быть использован для синтеза жиров.
В результате действия ЛП-липазы на ХМ количество жиров в них снижается на 90%, уменьшаются размеры частиц, апопротеин С-П переносится обратно на ЛПВП. Образовавшиеся частицы называются остаточными ХМ. Они содержат в себе фосфолипиды, холестерол, жирорастворимые витамины и апопротеины В-48 и Е. Остаточные ХМ захватываются гепатоцитами, которые имеют рецепторы, взаимодействующие с этими апопротеинами. Путём эндоцитоза остаточные ХМ попадают внутрь клеток, и ферментами лизосом белки и липиды гидролизуются, а затем утилизируются. Жирорастворимые витамины и экзогенный холестерол используются в печени или транспортируются в другие ткани. Гиперхиломикронемия, гипертриглицеролемия. После приёма пищи, содержащей жиры, развивается физиологическая гипертриглицеролемия и, соответственно, гиперхиломикронемия, которая может продолжаться до нескольких часов.
Скорость удаления ХМ из кровотока зависит от:
активности ЛП-липазы;
присутствия ЛПВП, поставляющих апопротеины С-II и Е для ХМ;
активности переноса апоС-II и апоЕ на ХМ.
Генетические дефекты любого из белков, участвующих в метаболизме ХМ, приводят к развитию семейной гиперхиломикронемии - гиперлипопротеинемии типа I. У таких больных в постабсорбтивном периоде концентрация триацилглицеролов повышена (более 200 мг/дл), плазма крови по виду напоминает молоко и при оставлении на холоде (+4 °С) в ней всплывают белые жирные хлопья, что характерно для гипертриглицеролемии и гиперхиломикронемии.
В тяжёлых случаях при этом заболевании происходит отложение триацилглицеролов в коже и сухожилиях в виде ксантом, у пациентов рано нарушается память, появляются боли в животе из-за сужения просвета сосудов и уменьшения кровотока, нарушается функция поджелудочной железы, что часто бывает причиной смерти больных. Если концентрация триацилглицеролов в крови превышает 4000 мг/дл, то липиды откладываются в сетчатке глаза, однако это не всегда влияет на зрительную функцию. При лечении гиперхиломикронемий необходимо прежде всего снизить потребление жиров с пищей, так как ХМ транспортируют экзогенные жиры
66-67 оксиление глицерина в тканях , ВЖК
Окисление глицерина и высших жирных к-т. Последовательность реакций. Связь β-окисления с циклом Кребса и дых цепью. Физиологическое значение окисления жирных кислот в зависимости от ритма питания и мышечной активности.
Д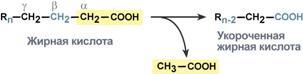 ля
преобразования энергии, заключенной в
жирных кислотах, в энергию связей АТФ
существует метаболический путь окисления
жирных кислот до СО2 и воды, тесно
связанный с циклом трикарбоновых кислот
и дыхательной цепью. Этот путь называется
β-окисление, т.к. происходит окисление
3-го углеродного атома жирной кислоты
(β-положение) в карбоксильную группу,
одновременно от кислоты отщепляется
ацетильная группа, включающая С1 и С2
исходной жирной кислоты. Реакции
β-окисления происходят в митохондриях
большинства клеток организма (кроме
нервных клеток). Для окисления используются
жирные кислоты, поступающие в цитозоль
из крови или появляющиеся при липолизе
собственных внутриклеточных ТАГ.
Суммарное уравнение окисления
пальмитиновой кислоты выглядит следующим
образом: Пальмитоил-SКоА + 7ФАД + 7НАД+ +
7Н2O + 7HS-KoA → 8Ацетил-SКоА + 7ФАДН2 + 7НАДН
ля
преобразования энергии, заключенной в
жирных кислотах, в энергию связей АТФ
существует метаболический путь окисления
жирных кислот до СО2 и воды, тесно
связанный с циклом трикарбоновых кислот
и дыхательной цепью. Этот путь называется
β-окисление, т.к. происходит окисление
3-го углеродного атома жирной кислоты
(β-положение) в карбоксильную группу,
одновременно от кислоты отщепляется
ацетильная группа, включающая С1 и С2
исходной жирной кислоты. Реакции
β-окисления происходят в митохондриях
большинства клеток организма (кроме
нервных клеток). Для окисления используются
жирные кислоты, поступающие в цитозоль
из крови или появляющиеся при липолизе
собственных внутриклеточных ТАГ.
Суммарное уравнение окисления
пальмитиновой кислоты выглядит следующим
образом: Пальмитоил-SКоА + 7ФАД + 7НАД+ +
7Н2O + 7HS-KoA → 8Ацетил-SКоА + 7ФАДН2 + 7НАДН
Э тапы окисления жирных кислот: 1. Прежде, чем проникнуть в матрикс митохондрий и окислиться, жирная к-та должна активироваться в цитозоле. Это осуществляется присоединением к ней коэнзима А с обр-ем ацил-S-КоА. Ацил-S-КоА яв-ся высокоэнергетическим соед-ем. Необратимость реакции достигается гидролизом дифосфата на 2 молекулы фосфорной к-ты.
2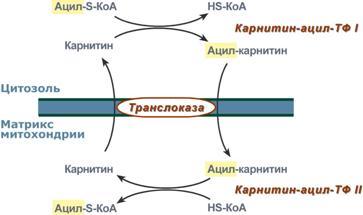 .
Ацил-S-КоА не способен проходить через
митохондриальную мембрану, поэтому
существует способ его переноса в
комплексе с витаминоподобным веществом
карнитином. На наружной мембране
митохондрий имеется фермент
карнитин-ацилтрансфераза I.
.
Ацил-S-КоА не способен проходить через
митохондриальную мембрану, поэтому
существует способ его переноса в
комплексе с витаминоподобным веществом
карнитином. На наружной мембране
митохондрий имеется фермент
карнитин-ацилтрансфераза I.
Карнитин синтезируется в печени и почках и затем транспортируется в остальные органы. Во внутриутробном периоде и в первые годы жизни значение карнитина для организма чрезвычайно важно. Энергообеспечение нервной системы детского организма и, в частности, головного мозга осуществляется за счет 2х параллельных процессов: карнитин-зависимого ок-ия жирных к-т и аэробного окисления глюкозы. Карнитин необходим для роста головного и спинного мозга, для вз-вия всех отделов НС, ответственных за движение и взаимодействие мышц. Сущ-ют исследования, связывающие с недостатком карнитина детский церебральный паралич и феномен "смерти в колыбели".
3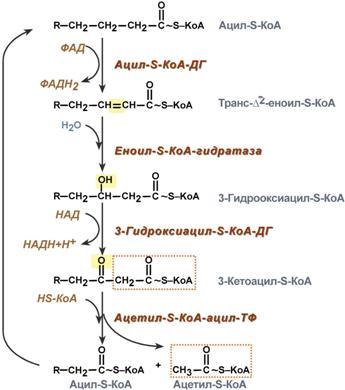 .
После св-ия с карнитином ж к-та переносится
ч/з мембрану транслоказой. Здесь на
внутренней стороне мембраны фермент
карнитин-ацилтрансфераза II вновь обр-ет
ацил-S-КоА кот вступает на путь β-окисления.
.
После св-ия с карнитином ж к-та переносится
ч/з мембрану транслоказой. Здесь на
внутренней стороне мембраны фермент
карнитин-ацилтрансфераза II вновь обр-ет
ацил-S-КоА кот вступает на путь β-окисления.
4. Процесс собственно β-окисления состоит из 4-х реакций, повторяющихся циклически. В них последовательно происходит окисление (ацил-SКоА-дегидрогеназа), гидратирование (еноил-SКоА-гидратаза) и вновь окисление 3-го атома углерода (гидроксиацил-SКоА-дегидрогеназа). В последней, трансферазной, реакции от жирной кислоты отщепляется ацетил-SКоА. К оставшейся (укороченной на два углерода) жирной кислоте присоединяется HS-КоА, и она возвращается к первой реакции. Все повторяется до тех пор, пока в последнем цикле не образуются два ацетил-SКоА.
Расчет энергетического баланса β-окисления: При расчете количества АТФ, образуемого при β-окислении жирных кислот необходимо учитывать:
*количество образуемого ацетил-SКоА – определяется обычным делением числа атомов углерода в жирной кислоте на 2;
* число
циклов β-окисления. Число циклов
β-окисления легко определить исходя из
представления о жирной кислоте как о
цепочке двухуглеродных звеньев. Число
разрывов между звеньями соответствует
числу циклов β-окисления. Эту же величину
можно подсчитать по формуле (n/2 -1), где
n – число атомов углерода в кислоте,
число
циклов β-окисления. Число циклов
β-окисления легко определить исходя из
представления о жирной кислоте как о
цепочке двухуглеродных звеньев. Число
разрывов между звеньями соответствует
числу циклов β-окисления. Эту же величину
можно подсчитать по формуле (n/2 -1), где
n – число атомов углерода в кислоте,
* число
двойных связей в жирной кислоте. В первой
реакции β-окисления происходит образование
двойной связи при участии ФАД. Если
двойная связь в жирной кислоте уже
имеется, то необходимость в этой реакции
отпадает и ФАДН2 не образуется. Количество
необразованных ФАДН2 соответствует
числу двойных связей. Остальные реакции
цикла идут без изменений;
число
двойных связей в жирной кислоте. В первой
реакции β-окисления происходит образование
двойной связи при участии ФАД. Если
двойная связь в жирной кислоте уже
имеется, то необходимость в этой реакции
отпадает и ФАДН2 не образуется. Количество
необразованных ФАДН2 соответствует
числу двойных связей. Остальные реакции
цикла идут без изменений;
*количество энергии АТФ, потраченной на активацию (всегда соответствует двум макроэргическим связям).
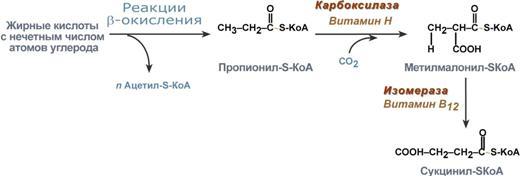
Окисление жирных кислот с нечетным числом углеродных атомов. Ж к-ты с нечетным числом углеродов поступают в организм с растительной пищей и морепродуктами. Их окисление происходит по обычному пути до последней реакции, в которой обр-ся пропионил-SКоА. Суть превращений пропионил-SКоА сводится к его карбоксилированию, изомеризации и обр-ию сукцинил-SКоА. В этих реакциях участвуют биотин и В12.
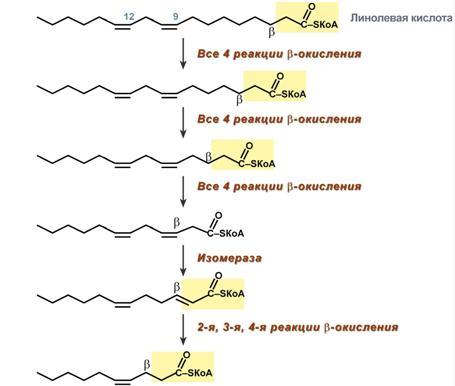
Окисление ненасыщенных жирных кислот. При окислении ненасыщенных жирных кислот возникает потребность клетки в дополнительных ферментах изомеразах. Эти изомеразы перемещают двойные связи в жирнокислотных остатках из γ- в β-положение и переводят природные двойные связи из цис- в транс-положение. Т.о., уже имеющаяся двойная связь готовится к β-окислению и пропускается первая реакция цикла, в которой участвует ФАД.
Глицерин – трехатомный спирт, водорастворим и легко всасывается из кишечника и по воротной вене поступает в печень. Окисление глицерина
Глицерин сначала фосфорилируется с участием АТФ до глицерофосфата (3-фосфоглицерол). Затем под действием НАД-зависимой глицерофосфатдегидрогеназы окисляется до 3-фосфоглицеринового альдегида. Фосфоглицериновый альдегид далее может окисляться до пировиноградной и молочной кислоты.
Связь в-окисления с ЦК и Дц: Таким образом, молекула жирной кислоты в конце концов распадается до продуктов, имеющих всего два углеродных атома, превращающихся в цикле трикарбоновых кислот. Восстановленные коферменты впоследствии вновь окисляются в дыхательной цепи с одновременным образованием макроэргических фосфатов. С точки зрения образования АТР, окисление жирных кислот составляет основной энергетический резерв организма.
Регуляция в-окисления: Ключевой фермент – карнитинацилтрансфераза1, аллостерический фермент, в печени его аллостерический ингибитор – малонилКоА. Активируют: катехоламины, СТГ, глюкагон. Ингибирует: инсулин.
Е значение в-окисление имеет для скелетных мышц (50% Е), для сердечной мышцы (70%), головной мозг и другие нервные ткани, а также эритроциты не используют жирные к-ты для окисления; они не поступают в головной мозг, т.к. не проходят ч/з гематоэнцефалический барьер.
68 Метаболизм ацетил КОА. Биосинтез Кетоновых тел
А![]()
 цетилКоА-
это центральный метаболит липидного
обмена.
цетилКоА-
это центральный метаболит липидного
обмена.
Источники: 1)Глюкоза 2)глицерин 3)АК} (при кратковременной напряженной мышечной работе) 4)Жирные к-ты (в-окисление при длительной мышечной работе, голодании, на холоде, при беременности и сахарном диабете). Пути использования образовавшегося ацетилКоА зависят от функционального состояния клетки (энергетический заряд) и ее специфики. Если в кл достаточно АТФ, то он используется на синтез ж к-т,т.к. АТФ активирует ключевой фермент ситеза ж к-т, а их накопление стимулирует синтез жира. Распад жира тормозится и в-окисление при этом тоже тормозится. Напряженная мышечная работа, стресс, увел-ие секреции катехоламинов активирует липолиз, в-окисление ж к-т; в этом случае актив-ся синтез кетоновых тел и ЦТК.
Пути использования: 1. окисляется в цикле Кребса(90%); 2. используется в синтезе ЖК (9%) 3. Образование В-гидрокси-в-метилглутарилКоА (а из него либо холестерин, либо кетоновые тела -1%)
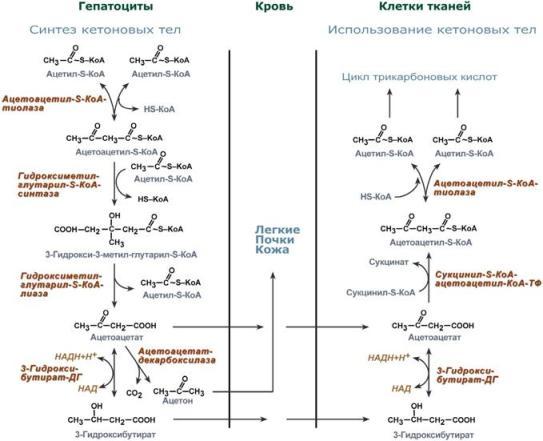
При состояниях, сопровождающихся снижением глюкозы крови, клетки органов и тканей испытывают энергетический голод. Так как окисление жирных кислот процесс "трудоемкий", а нервная ткань вообще неспособна окислять жирные кислоты, то печень облегчает использование этих кислот тканями, заранее окисляя их до уксусной кислоты и переводя последнюю в транспортную форму – кетоновые тела. К кетоновым телам относят три соединения близкой структуры – ацетоацетат, 3-гидроксибутират и ацетон. Стимулом для образования кетоновых тел служит поступление большого количества жирных кислот в печень. Как уже указывалось, при состояниях, активирующих липолиз в жировой ткани, не менее 30% образованных жирных кислот задерживаются печенью. К таким состояниям относится голодание, сахарный диабет I типа, длительные физические нагрузки. Так как синтез ТАГ в этих условиях невозможен, то жирные кислоты из цитозоля попадают в митохондрии и окисляются с образованием кетонов. Кроме отмеченных ситуаций, количество кетоновых тел в крови возрастает при алкогольном отравлении и потреблении жирной пищи. При богатой жирами диете, особенно у детей, жирные кислоты не успевают включиться в состав ТАГ и ЛПОНП и частично переходят в митохондрии, что увеличивает синтез кетоновых тел. При алкогольном отравлении субстратом для синтеза кетонов является ацетил-SКоА, синтезируемый при обезвреживании этанола. В обычных условиях синтез кетоновых тел также идет, хотя в гораздо меньшем количестве. Для этого используются как жирные кислоты, так и безазотистые остатки кетогенных и смешанных аминокислот. Синтез ацетоацетата происходит только в митохондриях печени, далее он либо восстанавливается до 3-гидроксибутирата, либо спонтанно декарбоксилируется до ацетона. Далее все три соединения поступают в кровь и разносятся по тканям. Ацетон, как летучее вещество, легко удаляется с выдыхаемым воздухом и потом. Все кетоновые тела могут выделяться с мочой. Используются кетоновые тела клетками всех тканей, кроме печени и эритроцитов. Особенно активно, даже в норме, они потребляются миокардом и корковым слоем надпочечников. Реакции утилизации кетоновых тел примерно совпадают с обратным направлением реакций синтеза. В цитозоле 3-гидроксибутират окисляется, образующийся ацетоацетат проникает в митохондрии, активируется за счет сукцинил-SКоА и превращается в ацетил-SКоА, который сгорает в ЦТК.
Регуляция синтеза кетоновых тел. Регуляторный фермент синтеза кетоновых тел - ГМГ-КоА синтаза.
*ГМГ-КоА-синтаза - индуцируемый фермент; его синтез увеличивается при повышении концентрации жирных кислот в крови. Концентрация жирных кислот в крови увеличивается при мобилизации жиров из жировой ткани под действием глюкагона, адреналина, т.е. при голодании или физической работе.
*ГМГ-КоА-синтаза ингибируется высокими концентрациями свободного кофермента А.
*Когда поступление жирных кислот в клетки печени увеличивается, КоА связывается с ними, концентрация свободного КоА снижается, и фермент становится активным.
*Если поступление жирных кислот в клетки печени уменьшается, то, соответственно, увеличивается концентрация свободного КоА, ингибирующего фермент. Следовательно, скорость синтеза кетоновых тел в печени зависит от поступления жирных кислот.
Кетоновые тела образуются в печени и выполняют следующие функции:1. Энергетическая. Скелетная и сердечная мышцы, мозг и др внепеченочные ткани обеспечивают энергетические потребности за счет катаболизма кетоновых тел. Печень не может окислять кетоновые тела. 2.необходимы для образования миелиновых оболочек нервов и белого вещества головного мозга.
Утилизация кетоновых тел происходит в митохондриях. Бета-гидроксибутират превращается в ацетоацетат, а ацетоацетат вступает в реакцию с промежуточным продуктом ЦТК - сукцинил-КоА. При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе. β-Гидроксибутират (рис. 8-34), попадая в клетки, дегидрируется NAD-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сук-цинил-КоА - донором КоА: Ацетоацетат + Сукцинил-КоА → Ацетоацетил- КоА + Сукцинат
Реакцию катализирует сукцинил-КоА-ацето-ацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не использует кетоновые тела как источники энергии, а производит их "на экспорт". Кетоновые тела - хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СО2 и Н2О обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата - 26 молекул.
В норме процессы синтеза и использования кетоновых тел уравновешены, поэтому концентрация кетоновых тел в крови и в тканях обычно очень низка, и составляет 0,12-0,30 ммоль/л.. В норме в крови кол-во КТ 1-3 мг, в моче 40мг. При сахарном диабете 10-50 мг в моче. Накопление кетоновых тел в организме называется кетозом.Он сопровождается кетонемией и кетонурией. Физиологиеский кетоз – при голодании, тяжелой мышечной работе, у новорожденных. Патологический – при сахарном диабете. Однако при общем или при углеводном голодании может нарушаться баланс между образованием и утилизацией кетоновых тел. Это связано с тем, что скорость образования кетоновых тел зависит от скорости -окисления жирных кислот в печени, а процесс -окисления ускоряется при усилении липолиза (распада жира) в жировой ткани. Усиление липолиза может происходить под действием гормона адреналина, при мышечной работе, при голодании. При недостатке инсулина (сахарный диабет) также происходит усиление липолиза. При усилении липолиза увеличивается скорость утилизации кетоновых тел, которые являются важными источниками энергии при мышечной работе, голодании.
Постепенное истощение запасов углеводов при сахарном диабете приводит к относительному отставанию утилизации кетоновых тел от кетогенеза. Причина отставания: не хватает сукцинил-КоА и ЩУК, которые, в основном, являются продуктом обмена углеводов. Поэтому верно выражение: "Жиры сгорают в пламени углеводов". Это означает, что для эффективного использования продуктов распада жира необходимы продукты углеводного обмена: сукцинил-КоА и ЩУК.
Т.о., при углеводном голодании концентрация кетоновых тел в крови увеличивается. На 3-й день голодания концентрация кетоновых тел в крови будет примерно 2-3 ммоль/л, а при дальнейшем голодании - гораздо более высокой. Это состояние называют гиперкетонемия. У здоровых людей при мышечной работе и при голодании наблюдается гиперкетонемия, но она незначительна.
Похожая ситуация характерна для сахарного диабета. При сахарном диабете клетки постоянное сильнейшее углеводное голодание, потому что глюкоза плохо проникает в клетки. Наблюдается активация липолиза и повышается образование кетоновых тел. При тяжелых формах сахарного диабета концентрация кетоновых тел в крови может быть еще выше, и достигать опасных для жизни значений: до 20 ммоль/л и более. Все кетоновые тела являются органическими кислотами. Их накопление приводит к сдвигу pH в кислую сторону. В клинике повышение концентрации кетоновых тел в крови называется гиперкетонемия, а сдвиг pH при этом в кислую сторону - кетоацидоз. Нарушается работа многих ферментативных систем. Увеличение концентрации ацетоацетата приводит к ускоренному образованию ацетона. Ацетон - токсичное вещество (органический растворитель). Он растворяется в липидных компонентах клеточных мембран и дезорганизует их. Страдают все ткани организма, а больше всего - клетки нервной ткани. Это может проявляться потерей сознания (гипергликемическая кома). В очень тяжелых случаях может наступить гибель организма. Организм пытается защититься, поэтому часть кетоновых тел удаляется с мочой. Появление кетоновых тел в моче - это кетонурия. Для распознавания гипер- и гипогликемической комы применяется экспресс-диагностика кетоновых тел. Основана на том, что гиперкетонемия приводит к выведению кетоновых тел с мочой (кетонурия). Поэтому проводят цветную реакцию на наличие кетоновых тел в моче. Раньше диагноз ставили по запаху ацетона изо рта больного при гипергликемической коме (запах гнилых яблок).
69 Биосинтез ЖК
Биосинтез жирных кислот наиболее активно происходит в цитозоле клеток печени, кишечника, жировой ткани в состоянии покоя или после еды.
Условно можно выделить 4 этапа биосинтеза:
1. Образование ацетил-SКоА из глюкозы, других моносахаров или кетогенных аминокислот.
2. Перенос ацетил-SКоА из митохондрий в цитозоль:*может быть в комплексе с карнитином, подобно тому как переносятся внутрь митохондрии высшие жирные кислоты, но здесь транспорт идет в другом направлении, *обычно в составе лимонной кислоты, образующейся в первой реакции ЦТК.
П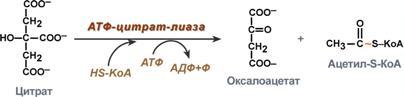
 оступающий
из митохондрий цитрат в цитозоле
расщепляется АТФ-цитрат-лиазой до
оксалоацетата и ацетил-SКоА. Оксалоацетат
в дальнейшем восстанавливается до
малата, и последний либо переходит в
митохондрии (малат-аспартатный челнок),
либо декарбоксилируется в
пируватмалик-ферментом ("яблочный"
фермент).
оступающий
из митохондрий цитрат в цитозоле
расщепляется АТФ-цитрат-лиазой до
оксалоацетата и ацетил-SКоА. Оксалоацетат
в дальнейшем восстанавливается до
малата, и последний либо переходит в
митохондрии (малат-аспартатный челнок),
либо декарбоксилируется в
пируватмалик-ферментом ("яблочный"
фермент).
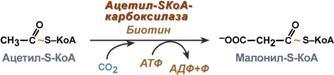
3. Образование малонил-SКоА из ацетил-SКоА. Карбоксилирование ацетил-SКоА катализируется ацетил-SКоА-карбоксилазой, мульферментным комплексом из трех ферментов.
4. Синтез пальмитиновой кислоты. Осуществляется мультиферментным комплексом "синтаза жирных кислот" (синоним пальмитатсинтаза) в состав которого входит 6 ферментов и ацил-переносящий белок (АПБ). Ацил-переносящий белок включает производное пантотеновой кислоты – 6-фосфопантетеин (ФП), имеющий HS-группу, подобно HS-КоА. Один их ферментов комплекса, 3-кетоацил-синтаза, также имеет HS-группу в составе цистеина. Взаимодействие этих групп обусловливает начало и продолжение биосинтеза жирной кислоты, а именно пальмитиновой кислоты. Для реакций синтеза необходим НАДФН. В первых двух реакциях последовательно присоединяются малонил-SКоА к фосфопантетеину ацил-переносящего белка и ацетил-SКоА к цистеину 3-кетоацилсинтазы. 3-Кетоацилсинтаза катализирует третью реакцию – перенос ацетильной группы на С2 малонила с отщеплением карбоксильной группы. Далее кетогруппа в реакциях восстановления (3-кетоацил-редуктаза), дегидратации (дегидратаза) и опять восстановления (еноил-редуктаза) превращается в метиленовую с образованием насыщенного ацила, связанного с фосфопантетеином. Ацилтрансфераза переносит полученный ацил на цистеин 3-кетоацил-синтазы, к фосфопантетеину присоединяется малонил-SКоА и цикл повторяется 7 раз до образования остатка пальмитиновой кислоты. После этого пальмитиновая кислота отщепляется шестым ферментом комплекса тиоэстеразой. Удлинение цепи жирных кислот
С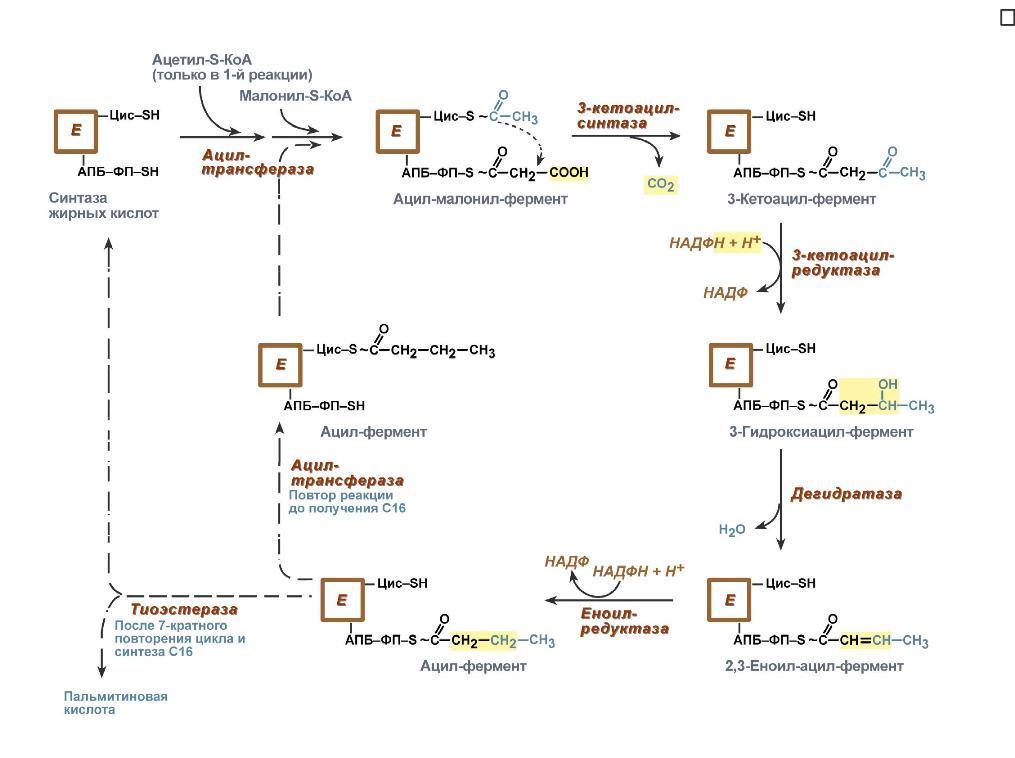 интезированная
пальмитиновая кислота при необходимости
поступает в эндоплазматический ретикулум
или в митохондрии. Здесь с участием
малонил-S-КоА и НАДФН цепь удлиняется
до С18 или С20. Удлиняться могут и
ненасыщенные жирные кислоты (олеиновая,
линолевая, линоленовая) с образованием
производных эйкозановой кислоты (С20).
Но двойная связь животными клетками
вводится не далее 9 атома углерода,
поэтому ω3- и ω6-полиненасыщенные жирные
кислоты синтезируются только из
соответствующих предшественников.
интезированная
пальмитиновая кислота при необходимости
поступает в эндоплазматический ретикулум
или в митохондрии. Здесь с участием
малонил-S-КоА и НАДФН цепь удлиняется
до С18 или С20. Удлиняться могут и
ненасыщенные жирные кислоты (олеиновая,
линолевая, линоленовая) с образованием
производных эйкозановой кислоты (С20).
Но двойная связь животными клетками
вводится не далее 9 атома углерода,
поэтому ω3- и ω6-полиненасыщенные жирные
кислоты синтезируются только из
соответствующих предшественников.
Синтезир-ся в орг-ме в основном пальмитиновая к-та. При необходимости ж к-ты с большим числом углеродных атомов. Ненасыщенные ж к-ты обр-ся на мембранах ЭПС с участием О2, НАДН и В5. Под воздействием ферментов десатураз обр-ся пальмитиновая и олеиновая к-ты. Полиненасыщенные ж к-ты (линолевая, арахидоновая, линоленовая) должны поступать с пищей. Источником углевода для синтеза ж к-т служит ацетилКоА, обр-ся при распаде углеводов. Избыток углеводов, поступающих в орг-м трансформир-ся в ж к-ты, а затем в жиры.
Лимитирующим ферментом является ацетил-КоАкарбоксилаза. Аллостерические активаторы — АТФ и цитрат, ингибиторы — жирные кислоты с длинной цепью. Инсулин, эстрогены активиру¬ют, катехоламины и стресс ингибируют синтез жирных кислот. Значение:при распаде УВ обр ацетил-Коа, который используется в синтезе ЖК, т.о. избыток УВ запасается в виде жира.