

Роль глюкозы в организме человека
Глюкоза – это основной источник энергии в организме. Клетки человеческого организма, расщепляя глюкозу, получают энергию, необходимую для поддержания жизнедеятельности. Глюкоза поступает в организм с пищей в составе углеводов (крахмал, сахар, мед и др.). Глюкоза содержится во всех сладких продуктах, в том числе и в сахаре. Нередко глюкозу ошибочно называют «сахаром» крови ( на самом деле сахар состоит из равных частей фруктозы и глюкозы и распадается на эти два элемента под действием специальных ферментов в кишечнике).
Регуляция уровня глюкозы (сахара) в крови.
Уровень глюкозы в крови варьирует в зависимости от приема пищи. После еды сахар крови всегда немного повышается, а затем нормализуется в течение нескольких часов. Повышение уровня глюкозы в крови после еды дает сигнал к выделению инсулина – гормона поджелудочной железы, способствующему усвоению глюкозы клетками организма и понижению ее концентрации в крови. Инсулин также способствует образованию запасов глюкозы в печени в виде гликогена. Параллельно с понижением уровня глюкозы в крови выделение инсулина уменьшается. Несмотря на хорошую растворимость в воде, у здоровых людей глюкоза не выводится вместе с мочой, потому что при нормальной концентрации глюкозы в крови почки успевают впитывать глюкозу из мочи обратно в кровь. При увеличении уровня глюкозы в крови выше определенного значения почки теряют способность впитывать глюкозу из мочи, что приводит к появлению глюкозурии (наличие глюкозы в моче). Глюкозурия характерна для сахарного диабета, однако, может указывать и на другие заболевания. Подробнее о регуляции уровня глюкозы в крови читайте в разделе Диабет
40)
Гликолиз
Гликолиз (от греч. glycys – сладкий и lysis – растворение, распад) – это последовательность ферментативных реакций, приводящих к превращению глюкозы в пируват с одновременным образованием АТФ.При аэробных условиях пируват проникает в митохондрии, где полностью окисляется до СО2 и Н2О. Если содержание кислорода недостаточно, как это может иметь место в активно сокращающейся мышце, пируват превращается в лактат.Итак, гликолиз – не только главный путь утилизации глюкозы в клетках, но и уникальный путь, поскольку он может использовать кислород, еслипоследний доступен (аэробные условия), но может протекать и в отсутствие кислорода (анаэробные условия).Анаэробный гликолиз – сложный ферментативный процесс распада глюкозы, протекающий в тканях человека и животных без потребления кислорода. Конечным продуктом гликолиза является молочная кислота. В процессе гликолиза образуется АТФ. Суммарное уравнение гликолиза можно представить следующим образом:
![]()
В анаэробных условиях гликолиз – единственный процесс в животном организме, поставляющий энергию. Именно благодаря гликолизу организм человека и животных определенный период может осуществлять ряд физиологических функций в условиях недостаточности кислорода. В тех случаях, когда гликолиз протекает в присутствии кислорода, говорят об аэробном гликолизе .Последовательность реакций анаэробного гликолиза, так же как и их промежуточные продукты, хорошо изучена. Процесс гликолиза катализируется одиннадцатью ферментами, большинство из которых выделено в гомогенном, клисталлическом или высокоочищенном виде и свойства которых достаточно известны. Заметим, что гликолиз протекает в гиало-плазме (цитозоле) клетки.Первой ферментативной реакцией гликолиза является фосфорили-рование, т.е. перенос остатка ортофосфата на глюкозу за счет АТФ. Реакция катализируется ферментом гексокиназой:
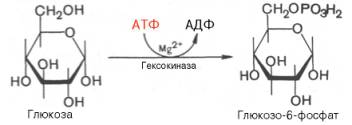
Образование глюкозо-6-фосфата в гексокиназной реакции сопровождается освобождением значительного количества свободной энергии системы и может считаться практически необратимым процессом.Наиболее важным свойством гексокиназы является ее ингибирование глюкозо-6-фосфатом, т.е. последний служит одновременно и продуктом реакции, и аллостерическим ингибитором.Фермент гексокиназа способен катализировать фосфорилирование не только D-глюкозы, но и других гексоз, в частности D-фруктозы, D-маннозы и т.д. В печени, кроме гексокиназы, существует фермент глюкокиназа, который катализирует фосфорилирование только D-глюкозы. В мышечной ткани этот фермент отсутствует (подробнее см. главу 16).Второй реакцией гликолиза является превращение глюкозо-6-фос-фата под действием фермента глюкозо-6-фосфат-изомеразы во фруктозо-6-фосфат:
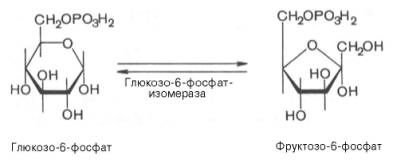
Эта реакция протекает легко в обоих направлениях, и для нее не требуется каких-либо кофакторов.
Третья реакция катализируется ферментом фосфофруктокиназой; образовавшийся фруктозо-6-фосфат вновь фосфорилируется за счет второй молекулы АТФ:
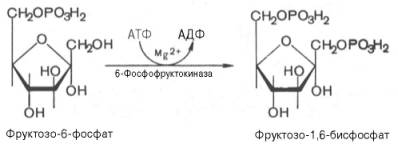
Данная реакция аналогично гексокиназной практически необратима, протекает в присутствии ионов магния и является наиболее медленно текущей реакцией гликолиза. Фактически эта реакция определяет скорость гликолиза в целом.Фосфофруктокиназа относится к числу аллостерических ферментов. Она ингибируется АТФ и стимулируется АМФ . При значительных величинах отношения АТФ/АМФ активность фосфофруктокиназы угнетается и гликолиз замедляется. Напротив, при снижении этого коэффициента интенсивность гликолиза повышается. Так, в неработающей мышце активность фосфофруктокиназы низкая, а концентрация АТФ относительно высокая. Во время работы мышцы происходит интенсивное потребление АТФ и активность фосфофруктокиназы повышается, что приводит к усилению процесса гликолиза.Четвертую реакцию гликолиза катализирует фермент альдолаза. Под влиянием этого фермента фруктозо-1,6-бисфосфат расщепляется на две фосфотриозы:
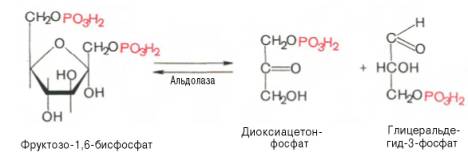
Эта реакция обратима. В зависимости от температуры равновесие устанавливается на различном уровне. При повышении температуры реакция сдвигается в сторону большего образования триозофосфатов (дигидро-ксиацетонфосфата и глицеральдегид-3-фосфата).Пятая реакция – это реакция изомеризации триозофосфатов. Катализируется ферментом триозофосфатизомеразой:

Равновесие данной изомеразной реакции сдвинуто в сторону дигид-роксиацетонфосфата: 95% дигидроксиацетонфосфата и около 5% глице-ральдегид-3-фосфата. В последующие реакции гликолиза может непосредственно включаться только один из двух образующихся триозофосфатов, а именно глицеральдегид-3-фосфат. Вследствие этого по мере потребления в ходе дальнейших превращений альдегидной формы фосфотриозы ди-гидроксиацетонфосфат превращается в глицеральдегид-3-фосфат.Образованием глицеральдегид-3-фосфата как бы завершается первая стадия гликолиза. Вторая стадия – наиболее сложная и важная. Она включает окислительно-восстановительную реакцию (реакция гликолитической оксидоредукции), сопряженную с субстратным фосфорилированием, в процессе которого образуется АТФ.В результате шестой реакции глицеральдегид-3-фосфат в присутствии фермента глицеральдегидфосфатдегидрогеназы, кофермента НАД и неорганического фосфата подвергается своеобразному окислению с образованием 1,3-бисфосфоглицериновой кислоты и восстановленной формы НАД (НАДН). Эта реакция блокируется йод- или бромацетатом, протекает в несколько этапов:
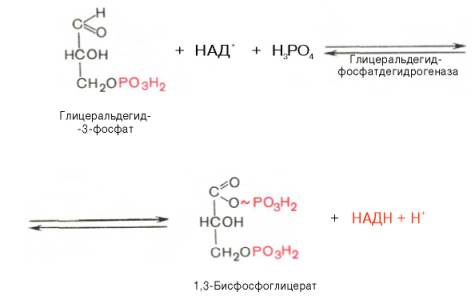
1,3-Бисфосфоглицерат представляет собой высокоэнергетическое соединение (макроэргическая связь условно обозначена знаком «тильда» ~). Механизм действия глицеральдегидфосфатдегидрогеназы сводится к следующему: в присутствии неорганического фосфата НАД+ выступает как акцептор водорода, отщепляющегося от глицеральдегид-3-фосфата. В процессе образования НАДН глицеральдегид-3-фосфат связывается с молекулой фермента за счет SH-групп последнего. Образовавшаяся связь богата энергией, но она непрочная и расщепляется под влиянием неорганического фосфата, при этом образуется 1,3-бисфосфоглицериновая кислота.Седьмая реакция катализируется фосфоглицераткиназой, при этом происходит передача богатого энергией фосфатного остатка (фосфатной группы в положении 1) на АДФ с образованием АТФ и 3-фосфогли-цериновой кислоты (3-фосфоглицерат):
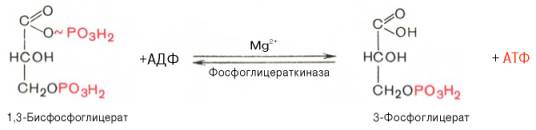
Таким образом, благодаря действию двух ферментов (глицеральде-гидфосфатдегидрогеназы и фосфоглицераткиназы) энергия, высвобождающаяся при окислении альдегидной группы глицеральдегид-3-фосфата до карбоксильной группы, запасается в форме энергии АТФ. В отличие от окислительного фосфорилирования образование АТФ из высокоэнергетических соединений называется субстратным фосфорилированием.Восьмая реакция сопровождается внутримолекулярным переносом оставшейся фосфатной группы, и 3-фосфоглицериновая кислота превращается в 2-фосфоглицериновую кислоту (2-фосфоглицерат).Реакция легкообратима, протекает в присутствии ионов Mg2+. Кофактором фермента является также 2,3-бисфосфоглицериновая кислота аналогично тому, как в фосфоглюкомутазной реакции роль кофактора выполняет глюкозо-1,6-бисфосфат:

Девятая реакция катализируется ферментом енолазой, при этом 2-фосфоглицериновая кислота в результате отщепления молекулы воды переходит в фосфоенолпировиноградную кислоту (фосфоенолпируват), а фосфатная связь в положении 2 становится высокоэргической:
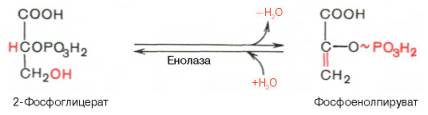
Енолаза активируется двухвалентными катионами Mg2+или Мn2+ и ингибируется фторидом.
Десятая реакция характеризуется разрывом высокоэргической связи и переносом фосфатного остатка от фосфоенолпирувата на АДФ (субстратное фосфорилирование). Катализируется ферментом пируваткиназой:
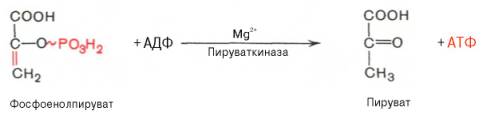
Для действия пируваткиназы необходимы ионы Mg2+, а также одновалентные катионы щелочных металлов (К+ или др.). Внутри клетки реакция является практически необратимой.В результате одиннадцатой реакции происходит восстановление пировиноградной кислоты и образуется молочная кислота. Реакция протекает при участии фермента лактатдегидрогеназы и кофермента НАДН, образовавшегося в шестой реакции:

Последовательность протекающих при гликолизе реакций представлена на рис. 10.3.
Реакция восстановления пирувата завершает внутренний окислительно-восстановительный цикл гликолиза. НАД+ при этом играет роль промежуточного переносчика водорода от глицеральдегид-3-фосфата (6-я реакция) на пировиноградную кислоту (11-я реакция), при этом сам он регенерируется и вновь может участвовать в циклическом процессе, получившем название гликолитический оксидоредукции.Биологическое значение процесса гликолиза заключается прежде всего в образовании богатых энергией фосфорных соединений. На первых стадиях гликолиза затрачиваются 2 молекулы АТФ (гексокиназная и фосфофрук-токиназная реакции). На последующих образуются 4 молекулы АТФ (фосфоглицераткиназная и пируваткиназная реакции). Таким образом, энергетическая эффективность гликолиза в анаэробных условиях составляет 2 молекулы АТФ на одну молекулу глюкозы.Как отмечалось, основной реакцией, лимитирующей скорость гликолиза, является фосфофруктокиназная. Вторая реакция, лимитирующая скорость и регулирующая гликолиз – гексокиназная реакция. Кроме того, контроль гликолиза осуществляется также ЛДГ и ее изоферментами.В тканях с аэробным метаболизмом (ткани сердца, почек и др.) преобладают изоферменты ЛДГ1 и ЛДГ2 (см. главу 4). Эти изоферменты инги-бируются даже небольшими концентрациями пирувата, что препятствует образованию молочной кислоты и способствует более полному окислению пирувата (точнее, ацетил-КоА) в цикле трикарбоновых кислот.В тканях человека, в значительной степени использующих энергию гликолиза (например, скелетные мышцы), главными изоферментами являются ЛДГ5 и ЛДГ4. Активность ЛДГ5 максимальна при тех концентрациях пирувата, которые ингибируют ЛДГ1. Преобладание изоферментов ЛДГ4 и ЛДГ5 обусловливает интенсивный анаэробный гликолиз с быстрым превращением пирувата в молочную кислоту.Как отмечалось, процесс анаэробного распада гликогена получил название гликогенолиза. Вовлечение D-глюкозных единиц гликогена в процесс гликолиза происходит при участии 2 ферментов – фосфорилазы а и фосфо-глюкомутазы. Образовавшийся в результате фосфоглюкомутазной реакции глюкозо-6-фосфат может включаться в процесс гликолиза. После образования глюкозо-6-фосфата дальнейшие пути гликолиза и гликогенолиза полностью совпадают:
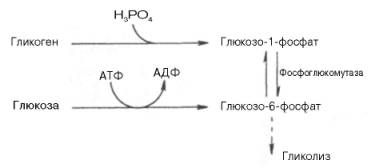
В процессе гликогенолиза в виде макроэргических соединений накапливаются не две, а три молекулы АТФ (АТФ не тратится на образование глюкозо-6-фосфата). Кажется, что энергетическая эффективность глико-генолиза выглядит несколько более высокой по сравнению с процессом гликолиза, но эта эффективность реализуется только при наличии активной фосфорилазы а. Следует иметь в виду, что в процессе активации фосфо-рилазы b расходуется АТФ (см. рис. 10.2).
41) Окисление глюкозы до СО2 и Н2О (аэробный распад). Аэробный распад глюкозы можно выразить суммарным уравнением:
С6Н12О6 + 6 О2 → 6 СО2 + Н2О + 2820 кДж/моль.
Этот процесс включает несколько стадий (рис. 7-33).
Аэробный гликолиз - процесс окисления глюкозы с образованием двух молекул пирувата;
Общий путь катаболизма, включающий превращение пирувата в ацетил-КоА и его дальнейшее окисление в цитратом цикле;
ЦПЭ на кислород, сопряжённая с реакциями дегидрирования, происходящими в процессе распада глюкозы.
В определённых ситуациях обеспечение кислородом тканей может не соответствовать их потребностям. Например, на начальных стадиях интенсивной мышечной работы при стрессе сердечные сокращения могут не достигать нужной частоты, а потребности мышц в кислороде для аэробного распада глюкозы велики. В подобных случаях включается процесс, который протекает без кислорода и заканчивается образованием лактата из пировиноградной кислоты. Этот процесс называют анаэробным распадом, или анаэробным гликолизом. Анаэробный распад глюкозы энергетически малоэффективен, но именно этот процесс может стать единственным источником энергии для мышечной клетки в описанной ситуации. В даньнейшем, когда снабжение мышц кислородом будет достаточным в результате перехода сердца на ускоренный ритм, анаэробный распад переключается на аэробный. Пути катаболизма глюкозы и их энергетический эффект показаны на рис
Аэробное окисление глюкозы В аэробных условиях глюкоза окисляется до СО2 и Н2О. Суммарное уравнение: С6Н12О6 + 6О2 → 6СО2+ 6Н2О + 2880 кДж/моль. Этот процесс включает несколько стадий: 1. Аэробный гликолиз. В нем происходит окисления 1 глюкозы до 2 ПВК, с образованием 2 АТФ (сначала 2 АТФ затрачиваются, затем 4 образуются) и 2 НАДН2; 2. Превращение 2 ПВК в 2 ацетил-КоА с выделением 2 СО2 и образованием 2 НАДН2; 3. ЦТК. В нем происходит окисление 2 ацетил-КоА с выделением 4 СО2, образованием 2 ГТФ (дают 2 АТФ), 6 НАДН2 и 2 ФАДН2; 4. Цепь окислительного фосфорилирования. В ней происходит окисления 10 (8) НАДН2, 2 (4) ФАДН2 с участием 6 О2, при этом выделяется 6 Н2О и синтезируется 34 (32) АТФ. В результате аэробного окисления глюкозы образуется 38 (36) АТФ, из них: 4 АТФ в реакциях субстратного фосфорилирования, 34 (32) АТФ в реакциях окислительного фосфорилирования. КПД аэробного окисления составит 65%. Анаэробное окисление глюкозы Катаболизм глюкозы без О2 идет в анаэробном гликолизе и ПФШ (ПФП). · В ходе анаэробного гликолиза происходит окисления 1 глюкозы до 2 молекул молочной кислоты с образованием 2 АТФ (сначала 2 АТФ затрачиваются, затем 4 образуются). В анаэробных условиях гликолиз является единственным источником энергии. Суммарное уравнение: С6Н12О6 + 2Н3РО4 + 2АДФ → 2С3Н6О3 + 2АТФ + 2Н2О. · В ходе ПФП из глюкозы образуются пентозы и НАДФН2. В ходе ПФШ из глюкозы образуются только НАДФН2. ГЛИКОЛИЗ Гликолиз – главный путь катаболизма глюкозы (а также фруктозы и галактозы). Все его реакции протекают в цитозоле. Аэробный гликолиз - это процесс окисления глюкозы до ПВК, протекающий в присутствии О2. Анаэробный гликолиз – это процесс окисления глюкозы до лактата, протекающий в отсутствии О2. Анаэробный гликолиз отличается от аэробного только наличием последней 11 реакции, первые 10 реакций у них общие. Этапы гликолиза В любом гликолизе можно выделить 2 этапа:
1 этап подготовительный, в нем затрачивается 2 АТФ. Глюкоза фосфорилируется и расщепляется на 2 фосфотриозы;
2 этап, сопряжён с синтезом АТФ. На этом этапе фосфотриозы превращаются в ПВК. Энергия этого этапа используется для синтеза 4 АТФ и восстановления 2НАДН2, которые в аэробных условиях идут на синтез 6 АТФ, а в анаэробных условиях восстанавливают ПВК до лактата.
42)
Метаболизм гликогена
Гликоген (см. с. 46) служит в животном организме резервом углеводов, из которого по мере метаболической потребности могут высвобождаться глюкозофосфат или глюкоза. Хранение в организме собственно глюкозы неприемлемо из-за ее высокой растворимости: высокие концентрации глюкозы создают в клетке высоко гипертоническую среду, что приводит к притоку воды. Напротив, нерастворимый гликоген осмотически почти неактивен.А. Метаболизм гликогенаГликоген животных, как и амилопектин растений, представляет собой разветвленный гомополимер глюкозы, в котором остатки глюкозы соединены α(1→4)-гликозидной связью. Связи в точках ветвления находятся в положении α(1→6) примерно каждого 10-го остатка. Таким образом, возникает древовидная структура с молекулярной массой >1ּ107 Да (до 50 000 остатков), в которой имеется только одна свободная аномерная ОН-группа, т. е. только один восстанавливающий конец.Гликоген печени никогда не расщепляется полностью. Как правило, укорачиваются или удлиняются (при высоком содержании глюкозы) только невосстанавливающие концы древовидной структуры. Удлинение цепи катализируется гликоген-синтазой [2]. Так как образование гликозидных связей между сахарами является эндоэргической реакцией, вначале в реакции глюкозо-1-фосфата с уридинтрифосфатом [УТФ (UTP)] образуется активированный предшественник — УДФ-глюкоза (UDP-глюкоза) ([1], см. с. 112). После этого остаток глюкозы легко переносится с этого промежуточного соединения на гликоген. Когда растущая цепь достигает определенной длины (>11 остатков), специальный фермент ветвления гликогена (1,4→1,6-трансгликозидаза ) [3] катализирует перенос концевого олигосахарида, состоящего из 6-7 остатков, на 6-ОН остаток глюкозы той же или другой цепи гликогена с образованием точки ветвления [α(1→6)-связи] Дальнейшее удлинение этого фрагмента осуществляется гликоген-синтазой, образующей α(1→4)-связи.Разветвленная структура гликогена облегчает быстрое освобождение углеводных остатков. Наиболее важным ферментом деградации гликогена является гликоген-фосфорилаза [4], отщепляющая от невосстанавливающего конца цепи остатки глюкозы в виде глюкозо-1-фосфата. Чем больше таких концов, тем больше молекул фосфорилазы могут действовать одновременно. Образование глюкозо-1-фосфата вместо глюкозы имеет то преимущество, что для включения освобожденных остатков глюкозы в гликолиз или ГМП не требуется АТФ.Благодаря структуре гликоген-фосфорилазы (см. с. 122), процесс последовательного отщепления останавливается за 4 остатка глюкозы от точки разветвления. Точки ветвления удаляются двумя другими ферментами [5 и 6]. Вначале трисахарид боковой цепи переносится [5] к невосстанавливающему концу главной цепи. Затем 1,6-гликозидаза [6] отщепляет остающийся единичный остаток глюкозы в точке ветвления в виде свободной глюкозы, после чего неразветвленная цепь, может вновь расщепляться фосфорилазой.Регуляция метаболизма гликогена путем взаимопревращений и роль гормонов рассмотрены на с. 122.Б. Баланс гликогенаВ организме человека может содержаться до 450 г гликогена, треть из которого накапливается в печени, а остальное — главным образом в мышцах. Содержание гликогена в других органах незначительно. Гликоген печени служит прежде всего для поддержания уровня глюкозы в крови в фазе пострезорбции (см. с. 300). Поэтому содержание гликогена в печени варьирует в широких пределах. При длительном голодании оно падает почти до нуля, после чего начинается снабжение организма глюкозой с помощью глюконеогенеза (см. с. 156). Гликоген мышц служит резервом энергии и не участвует в регуляции уровня глюкозы в крови. В мышцах отсутствует глюкозо-6-фосфатаза, поэтому гликоген мышц не может быть источником глюкозы в крови. По этой причине колебания содержания гликогена в мышцах меньше, чем в печени.
43) Окисление глицерина в тканях тесно связано с ГЛИКОЛИЗОМ, в который вовлекаются метаболиты обмена глицерина по следующей схеме: При окислении глицерина образовались конечные продукты: СО2 на этапе превращения: 1. ПИРУВАТА 2. ИЗОЦИТРАТА 3. Альфа-КЕТОГЛУТАРАТА Н2О на этапе превращения: 1 . альфа -ГЛИЦЕРОФОСФАТА 2. ГЛИЦЕРАЛЬДЕГИД-3-ФОСФАТА 3. 2-ФОСФОГЛИЦЕРАТА 4. ПИРУВАТА 5. ИЗОЦИТРАТА 6. Альфа-КЕТОГЛУТАРАТА 7. СУКЦИНАТА 8. МАЛАТА АТФ выделилось за счёт реакций А) Субстратного фосфорилиования на этапах превращения: 1. 1,3-дифосфоглицерата 2.2-фосфоенолпирувата 3.Сукцинил-КоА Б) Окислительного фосфорилирования на этапах превращения: 1.Альфа-глицерафосфата 2.Глицеральдегид-3 фосфата 3.Пирувата 4.Изоцитрата 5. Альфа-КЕТОГЛУТАРАТА 6.Сукцината 7.Малата
При окислении глицерина в зависимости от характера окислителя и условий окисления получаются различные соединения: глицериновый альдегид СН2ОНСНОНСНО, диоксиацетон СН2ОНСОСН2ОН, глицериновая кислота СН2ОНСНОН СООН, тартроновая кислота (СООН)2СНОН, мезоксалевая кислота (СООН)2СО; в кислом растворе KMnO4 или K2Cr2O7 окисляют глицерин до СО2 и воды.При действии на глицерин водоотнимающих средств или при нагревании его в присутствии щёлочи или сильных кислот образуются полиглицерины (ди-, три– и т.д.); при действии органических кислот, их ангидридов, хлорангидридов получаются сложные эфиры (глицериды) - моно-, ди– и триглицериды, например, 3RCOOH + C3H5(OH)3 → C3H5(OCOR)3 + 3H2O; при действии на глицерин минеральных кислот образуются сложные эфиры, например, с азотной кислотой в присутствии серной — тринитрат глицерина CH2ONO2 — CHONO2 — CH2ONO; с водоотнимающими средствами, например, KHSO4, даёт акролеин СН2 = СНСНО; при конденсации глицерина с альдегидами или кетонами — ацетали или кетали (например, глицеринацетальдегид, глицеринацетон и другие). Глицерин является предшественником для синтеза триацилглицеридов и фосфолипидов в печени и жировой ткани. Когда в качестве источника энергии организм использует запасенные жиры, глицерин и жирные кислоты попадают в кровь. Перед тем, как глицерин вступит в процесс гликолиза, его необходимо превратить в промежуточный продукт глицеральдегид-3-фосфат).Таким образом, глицерин сначала фосфорилируется с участием АТФ до глицерофосфата (3-фосфоглицерол)
44) Установлено, что окисление жирных кислот протекает в печени, почках, скелетных и сердечной мышцах, в жировой ткани. В мозговой ткани скорость окисления жирных кислот весьма незначительна; основным источником энергии в мозговой ткани служит глюкоза.
В 1904 г. Ф. Кнооп (F. Knoop) выдвинул гипотезу β-окисления жирных кислот на основании опытов по скармливанию собакам различных жирных кислот, в которых один атом водорода в концевой метильной группе (ω-углеродного атома) был замещен радикалом (С6Н5–).Ф. Кнооп высказал предположение, что окисление молекулы жирной кислоты в тканях организма происходит в β-положении. В результате от молекулы жирной кислоты последовательно отщепляются двууглеродные фрагменты со стороны карбоксильной группы.Жирные кислоты, входящие в состав естественных жиров животных и растений, имеют четное число углеродных атомов. Любая такая кислота, от которой отщепляется по паре углеродных атомов, в конце концов проходит через стадию масляной кислоты. После очередного β-окисления масляная кислота становится ацетоуксусной. Последняя затем гидроли-зуется до двух молекул уксусной кислоты. Теория β-окисления жирных кислот, предложенная Ф. Кноопом, в значительной мере послужила основой современных представлений о механизме окисления жирных кислот.Доставка жирных кислот к месту их окисления – к митохондриям – происходит сложным путем: при участии альбумина осуществляется транспорт жирной кислоты в клетку; при участии специальных белков (fatty acid binding proteins, FABP) – транспорт в пределах цитозоля; при участии карнитина – транспорт жирной кислоты из цитозоля в митохондрии.Процесс окисления жирных кислот складывается из следующих основных этапов.Активация жирных кислот. Свободная жирная кислота независимо от длины углеводородной цепи является метаболически инертной и не может подвергаться никаким биохимическим превращениям, в том числе окислению, пока не будет активирована. Активация жирной кислоты протекает на наружной поверхности мембраны митохондрий при участии АТФ, коэнзима A (HS-KoA) и ионов Mg2+. Реакция катализируется ферментом ацил-КоА-синтетазой:
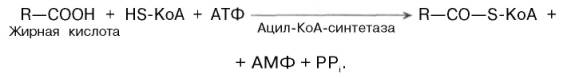
В результате реакции образуется ацил-КоА, являющийся активной формой жирной кислоты.Считают, что активация жирной кислоты протекает в 2 этапа. Сначала жирная кислота реагирует с АТФ с образованием ациладенилата, представляющим собой эфир жирной кислоты и АМФ. Далее сульфгидрильная группа КоА действует на прочно связанный с ферментом ациладенилат с образованием ацил-КоА и АМФ.Транспорт жирных кислот внутрь митохондрий. Коэнзимная форма жирной кислоты, в равной мере как и свободные жирные кислоты, не обладает способностью проникать внутрь митохондрий, где, собственно, и протекает их окисление. Переносчиком активированных жирных кислотс длинной цепью через внутреннюю митохондриальную мембрану служит карнитин. Ацильная группа переносится с атома серы КоА на гидро-ксильную группу карнитина с образованием ацилкарнитина, который диффундирует через внутреннюю митохондриальную мембрану:
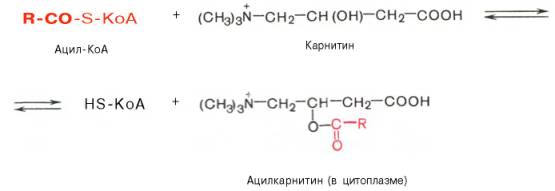
Реакция протекает при участии специфического цитоплазматического фермента карнитин-ацилтрансферазы. Уже на той стороне мембраны, которая обращена к матриксу, ацильная группа переносится обратно на КоА, что термодинамически выгодно, поскольку О-ацильная связь в кар-нитине обладает высоким потенциалом переноса группы. Иными словами, после прохождения ацилкарнитина через мембрану митохондрий происходит обратная реакция – расщепление ацилкарнитина при участии HS-KoA и митохондриальной карнитин-ацилтрансферазы:
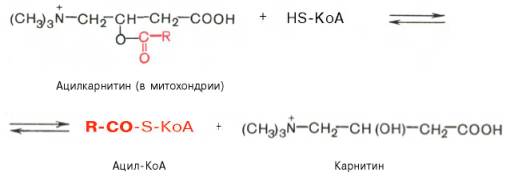
Внутримитохондриальное окисление жирных кислот. Процесс окисления жирной кислоты в митохондриях клетки включает несколько последовательных энзиматических реакций.Первая стадия дегидрирования. Ацил-КоА в митохондриях прежде всего подвергается ферментативному дегидрированию, при этом ацил-КоА теряет 2 атома водорода в α- и β-положениях, превращаясь в КоА-эфир ненасыщенной кислоты. Таким образом, первой реакцией в каждом цикле распада ацил-КоА является его окисление ацил-КоА-де-гидрогеназой, приводящее к образованию еноил-КоА с двойной связью между С-2 и С-3:
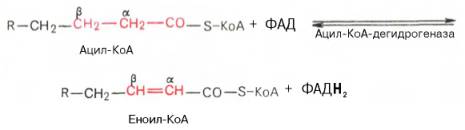
Существует несколько ФАД-содержащих ацил-КоА-дегидрогеназ, каждая из которых обладает специфичностью по отношению к ацил-КоА с определенной длиной углеродной цепи.
Стадия гидратации. Ненасыщенный ацил-КоА (еноил-КоА) при участии фермента еноил-КоА-гидратазы присоединяет молекулу воды. В результате образуется β-оксиацил-КоА (или 3-гидроксиацил-КоА):
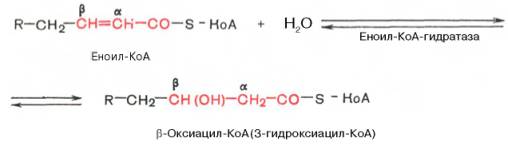
Заметим, что гидратация еноил-КоА стереоспецифична, подобно гидратации фумарата и аконитата (см. с. 348). В результате гидратации транс-Δ2-двойной связи образуется только L-изомер 3-гидроксиацил-КоА.Вторая стадия дегидрирования. Образовавшийся β-оксиацил-КоА (3-гидроксиацил-КоА) затем дегидрируется. Эту реакцию катализируют НАД+-зависимые дегидрогеназы:
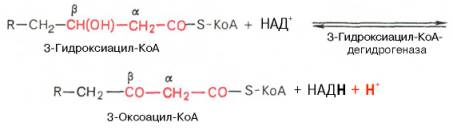
Тиолазная реакция. В ходе предыдущих реакций происходило окисление метиленовой группы при С-3 в оксогруппу. Тиолазная реакция представляет собой расщепление 3-оксоацил-КоА с помощью тиоловой группы второй молекулы КоА. В результате образуется укороченный на два углеродных атома ацил-КоА и двууглеродный фрагмент в виде ацетил-КоА. Данная реакция катализируется ацетил-КоА-ацилтрансферазой (β-ке-тотиолазой):
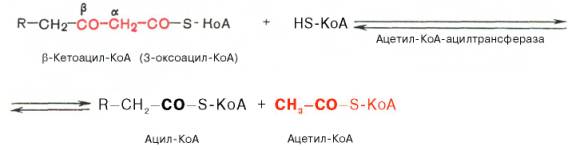
Образовавшийся ацетил-КоА подвергается окислению в цикле трикар-боновых кислот, а ацил-КоА, укоротившийся на два углеродных атома, снова многократно проходит весь путь β-окисления вплоть до образования бутирил-КоА (4-углеродное соединение), который в свою очередь окисляется до 2 молекул ацетил-КоА (рис. 11.2). Например, при окислении пальмитиновой кислоты (С16) повторяется 7 циклов β-окисления. Запомним, что при окислении жирной кислоты, содержащей п углеродных
атомов, происходит n/2–1 цикл β-окисления (т.е. на один цикл меньше, чем n/2, так как при окислении бутирил-КоА сразу происходит образование 2 молекул ацетил-КоА) и всего получится п/2 молекул ацетил-КоА. Следовательно, суммарное уравнение β-окисления активированной кислоты можно записать так:
Пальмитоил-КоА + 7ФАД + 7НАД+ + 7Н2O + 7HS-KoA –>
–> 8Ацетил-КоА + 7ФАДН2 + 7НАДН + 7Н+.
Баланс энергии. При каждом цикле β-окисления образуются одна молекула ФАДН2 и одна молекула НАДН. Последние в процессе окисления в дыхательной цепи и сопряженного с ним фосфорилирования дают: ФАДН2 – 2 молекулы АТФ и НАДН – 3 молекулы АТФ, т.е. в сумме за один цикл образуется 5 молекул АТФ. При окислении пальмитиновой кислоты образуется 5 х 7 = 35 молекул АТФ. В процессе β-окисления пальмитиновой кислоты образуется 8 молекул ацетил-КоА, каждая из которых, «сгорая» в цикле трикарбоновых кислот, дает 12 молекул АТФ, а 8 молекул ацетил-КоА дадут 12 х 8 = 96 молекул АТФ.Таким образом, всего при полном β-окислении пальмитиновой кислоты образуется 35 + 96 = 131 молекула АТФ. С учетом одной молекулы АТФ, потраченной в самом начале на образование активной формы пальмитиновой кислоты (пальмитоил-КоА), общий энергетический выход при полном окислении одной молекулы пальмитиновой кислоты в условиях животного организма составит 131 – 1 = 130 молекул АТФ. Изменение свободной энергии ΔF при полном сгорании 1 моля пальмитиновой кислоты составляет 2338 ккал, а богатая энергией фосфатная связь АТФ характеризуется величиной 7,6 ккал/моль. Нетрудно подсчитать, что примерно 990 ккал (7,6 х 130), или 42% от всей потенциальной энергии пальмитиновой кислоты при ее окислении в организме, используется для ресинтеза АТФ, а оставшаяся часть, очевидно, теряется в виде тепла.Следовательно, эффективность накопления энергии в результате окисления жирных кислот при стандартных условиях составляет ~ 40%, что близко к соответствующей величине для гликолиза, цикла трикарбоновых кислот и окислительного фосфорилирования.
45) В настоящее время в достаточной степени изучен механизм биосинтеза жирных кислот в организме животных и человека, а также катализирующие этот процесс ферментные системы. Синтез жирных кислот протекает в цитоплазме клетки. В митохондриях в основном происходит удлинение существующих цепей жирных кислот. Установлено, что в цитоплазме печеночных клеток синтезируется пальмитиновая кислота (16 углеродных атомов), а в митохондриях этих клеток из уже синтезированной в цитоплазме клетки пальмитиновой кислоты или из жирных кислот экзогенного происхождения, т.е. поступающих из кишечника, образуются жирные кислоты, содержащие 18, 20 и 22 углеродных атома.Иными словами, митохондриальная система биосинтеза жирных кислот, включающая несколько модифицированную последовательность реакций β-окисления, осуществляет только удлинение существующих в организме среднецепочечных жирных кислот, в то время как полный биосинтез пальмитиновой кислоты из ацетил-КоА активно протекает в цитозоле, т.е. вне митохондрий, по совершенно другому пути.Внемитохондриальная система биосинтеза de novo жирных кислот (ли-погенез). Эта система находится в растворимой (цитозольной) фракции клеток многих органов, в частности печени, почек, мозга, легких, молочной железы, а также в жировой ткани. Биосинтез жирных кислот протекает с участием НАДФН, АТФ, Мn2+ и НСО3– (в качестве источника СО2); субстратом является ацетил-КоА, конечным продуктом – пальмитиновая кислота. Потребности в кофакторах процессов биосинтеза и β-окисления жирных кислот значительно различаются.Как отмечалось, строительным блоком для синтеза жирных кислот в цитозоле клетки служит ацетил-КоА, который в основном поступает из митохондрий. Было выявлено, что цитрат стимулирует синтез жирных кислот в цитозоле клетки. Известно также, что образующийся в митохондриях в процессе окислительного декарбоксилирования пирувата и окисления жирных кислот ацетил-КоА не может диффундировать в цито-золь клетки, так как митохондриальная мембрана непроницаема для данного субстрата. Поэтому вначале внутримитохондриальный ацетил-КоА взаимодействует с оксалоацетатом, в результате чего образуется цитрат. Реакция катализируется ферментом цитрат-синтазой. Образовавшийся цитрат переносится через мембрану митохондрий в цитозоль при помощи специальной трикарбоксилаттранспортирующей системы.В цитозоле цитрат реагирует с HS-KoA и АТФ, вновь распадаясь на ацетил-КоА и оксалоацетат. Эта реакция катализируется АТФ-цитрат-лиа-зой. Уже в цитозоле оксалоацетат при участии цитозольной малатдегидро-геназы восстанавливается до малата. Последний при помощи дикарбокси-латтранспортирующей системы возвращается в митохондриальный мат-рикс, где окисляется до оксалоацетата, завершая тем самым так называемый челночный цикл:
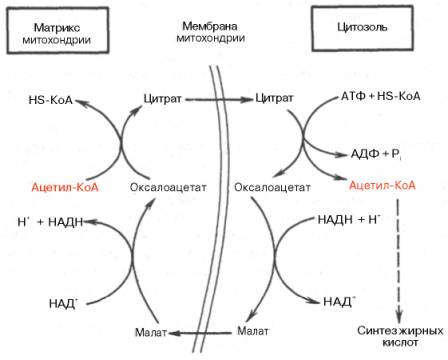
Существует еще один путь переноса внутримитохондриального аце-тил-КоА в цитозоль клетки – с участием карнитина. Как отмечалось, кар-нитин играет роль переносчика ацильных групп из цитозоля в митохондрии при окислении жирных кислот. По-видимому, он может выполнять эту роль и в обратном процессе, т.е. в переносе ацильных радикалов, в том числе ацетильного радикала, из митохондрий в цитозоль клетки. Однако, когда речь идет о синтезе жирных кислот, данный путь переноса ацетил-КоА не является главным.Образование малонил-КоА. Первой реакцией биосинтеза жирных кислот является карбоксилирование ацетил-КоА, для чего требуются бикарбонат, АТФ, ионы марганца. Катализирует эту реакцию фермент ацетил-КоА-кар-боксилаза. Фермент содержит в качестве простетической группы биотин. Авидин – ингибитор биотина угнетает эту реакцию, как и синтез жирных кислот в целом.
Установлено, что ацетил-КоА-карбоксилаза состоит из переменного числа одинаковых субъединиц, каждая из которых содержит биотин, биотинкарбоксилазу, карбоксибиотинпереносящий белок, транскарбоксилазу, а также регуляторный ал-лостерический центр, т.е. представляет собой полиферментный комплекс.Реакция протекает в два этапа: I – карбоксилирование биотина с участием АТФ и II – перенос карбоксильной группы на ацетил-КоА, в результате чего образуется малонил-КоА:
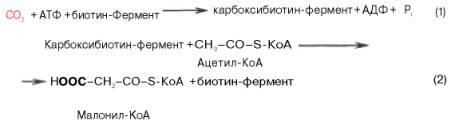
Малонил-КоА представляет собой первый специфический продукт биосинтеза жирных кислот. В присутствии соответствующей ферментной системы малонил-КоА быстро превращается в жирные кислоты.Энзиматические системы, осуществляющие синтез жирных кислот, называются жирно-кислотными синтетазами. Они широко встречаются в природе и могут быть изолированы из различных одноклеточных организмов, растений и животных тканей.Жирно-кислотные синтетазы делятся на 2 группы. К первой группе относятся полиэнзимные, не поддающиеся фракционированию комплексы с мол. м. порядка 500000, в которых все индивидуальные энзимы собраны в компактную структуру. В частности, в эту группу входят жирно-кислотные синтетазы животных тканей и дрожжей.Вторая группа включает жирно-кислотные синтетазы, из которых отдельные энзимы могут быть выделены методами белкового фракционирования. Такие синтетазы встречаются у ряда микроорганизмов (в частности, у E.coli) и растений. Иными словами, в этих случаях все индивидуальные ферменты синтетазной системы находятся в виде автономных полипептидов.Мультиферментный комплекс, называемый синтетазой (синтазой) жирных кислот, состоит из 6 ферментов, связанных с так называемым ацилпереносящим белком (АПБ). Этот белок относительно термостабилен, имеет две свободные HS-группы (цистеина и фосфопантетеинового остатка, присоединенного к ОН-группе серина) и вовлекается в процесс синтеза высших жирных кислот практически на всех его этапах. Мол. масса АПБ составляет около 10000. Данный белок в синтетазной системе выполняет роль КоА. Заметим, что в животных тканях не удалось обнаружить свободного АПБ, подобного микробному. Из печени выделен полиэнзимный комплекс, содержащий все энзимы, необходимые для синтеза жирных кислот. Энзимы комплекса настолько прочно связаны друг с другом, что все попытки изолировать их в индивидуальном виде не увенчались успехом. Приводим последовательность реакций, происходящих при синтезе жирных кислот:
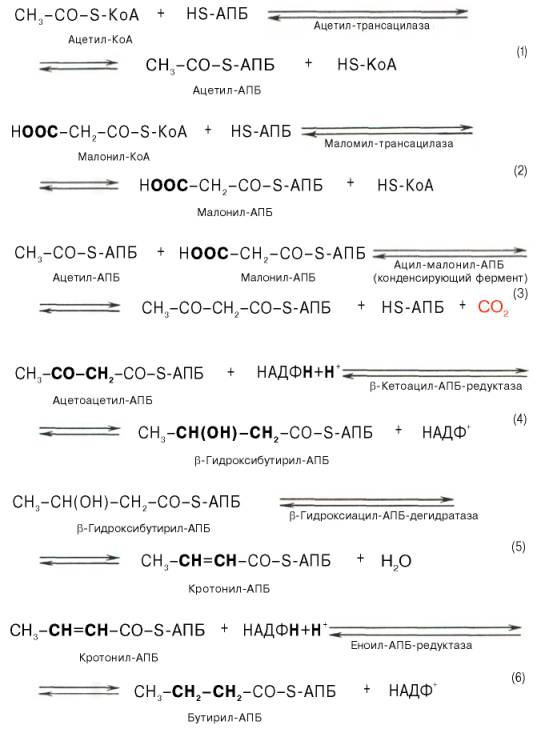
Далее цикл реакций повторяется. Допустим, что идет синтез пальмитиновой кислоты (С16). В этом случае образованием бутирил-АПБ завершается лишь первый из 7 циклов, в каждом из которых началом является присоединение молекулы малонил-АПБ к карбоксильному концу растущей цепи жирной кислоты. При этом отщепляется дистальная карбоксильная группа малонил-АПБ в виде СО2. Например, образовавшийся в первом цикле бутирил-АПБ взаимодействует с малонил-АПБ:
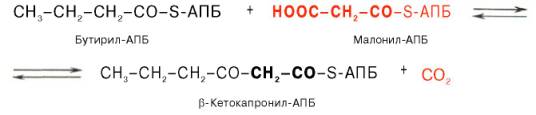
Завершается синтез жирной кислоты отщеплением HS-АПБ от ацил-АПБ под влиянием фермента деацилазы. Например:

Суммарное уравнение синтеза пальмитиновой кислоты можно записать так:
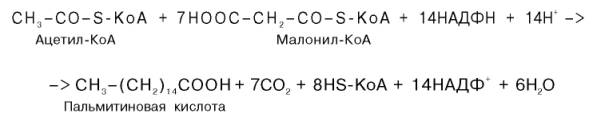
Или, учитывая, что на образование одной молекулы малонил-КоА из ацетил-КоА расходуются одна молекула АТФ и одна молекула СО2, которая затем отщепляется, суммарное уравнение можно представить в следующем виде:
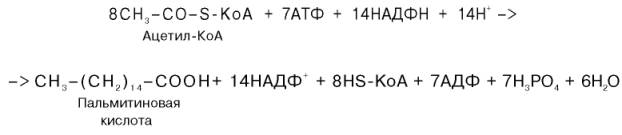
Основные этапы биосинтеза жирных кислот можно представить в виде схемы:В общем виде синтез жирных кислот у кишечной палочки представлен на рис. 11.4. Последовательность и характер реакций в синтезе жирных кислот, начиная с образования β-кетоацил-АПБ (на рис. 11.4 – ацетоацетил-АПБ) и кончая завершением одного цикла удлинения цепи на два углеродных атома, являются как бы обратными реакциями окисления жирных кислот. На самом деле пути синтеза и окисления жирных кислот не пересекаются даже частично. Это становится очевидным, если принять во внимание некоторые особенности синтеза и окисления жирных кислот.По сравнению с β-окислением биосинтез жирных кислот имеет ряд характерных особенностей: синтез жирных кислот в основном осуществляется в цитозоле клетки, а окисление – в митохондриях; участие в процессе биосинтеза жирных кислот малонил-КоА, который образуется путем связывания СО2 (в присутствии биотин-фермента и АТФ) с ацетил-КоА; на всех этапах синтеза жирных кислот принимает участие ацилпереносящий белок (HS-АПБ); при биосинтезе образуется D(–)-изомер 3-гидроксикис-лоты, а не L(+)-изомер, как это имеет место при β-окислении жирных кислот; необходимость для синтеза жирных кислот кофермента НАДФН. Последний в организме частично (на 50%) образуется в реакциях пен-тозофосфатного цикла, частично – в других реакциях, в частности в реакциях:
Малат + НАДФ+-> Пируват + С02 + НАДФН + Н+ Изоцитрат + НАДФ+-> α-Кетоглутарат + С02 + НАДФН + Н +.
Образование ненасыщенных жирных кислот. Элонгация жирных кислот. В отличие от растительных тканей ткани животных обладают весьма ограниченной способностью превращать насыщенные жирные кислоты в ненасыщенные.Установлено, что две наиболее распространенные мононенасыщенные жирные кислоты – пальмитоолеиновая и олеиновая – синтезируются из пальмитиновой и стеариновой кислот.Эти превращения протекают в микросомах клеток печени и жировой ткани при участии молекулярного кислорода, восстановленной системы пиридиновых нуклеотидов и цитохрома b5. Превращению подвергаются только активированные формы пальмитиновой и стеариновой кислот. Ферменты, участвующие в этих превращениях, получили название деса-тураз.Наряду с десатурацией жирных кислот (образование двойных связей) в микросомах происходит и их удлинение (элонгация), причем оба эти процесса могут сочетаться и повторяться. Удлинение цепи жирной кислоты происходит путем последовательного присоединения к соответствующему ацил-КоА двууглеродных фрагментов при участии малонил-КоА и НАДФН. Энзиматическая система, катализирующая удлинение жирных кислот, получила название элонгазы. На схеме представлены пути превращения пальмитиновой кислоты в реакциях десатурации и элонгации.Синтез жирных кислот протекает в цитоплазме клетки. В митохондриях в основном происходит удлинение существующих цепей жирных кислот. Установлено, что в цитоплазме пече-ночных клеток синтезируется пальмитиновая кислота (16 углеродных атомов), а в митохондриях этих клеток из уже синтезированной в цитоплазме клетки пальмитиновой кислоты или из жирных кислот экзогенного происхождения, т.е. поступающих из кишечника, образуются жирные кислоты, содержащие 18, 20 и 22 углеродных атома. Первой реакцией биосинтеза жирных кислот является карбоксилирование ацетил-КоА, для чего требуются бикарбонат, АТФ, ионы марганца. Катализирует эту реакцию фермент ацетил-КоА-кар-боксилаза. Фермент содержит в качестве простетической группы биотин. Реакция протекает в два этапа: I – карбоксилирование биотина с участием АТФ и II – перенос карбоксильной группы на ацетил-КоА, в результате чего образуется малонил-КоА. Малонил-КоА представляет собой первый специфический продукт биосинтеза жирных кислот. В присутствии соответствующей ферментной системы малонил-КоА быстро превращается в жирные кислоты.
46) Известно, что скорость биосинтеза жирных кислот во многом определяется скоростью образования триглицеридов и фосфолипидов, так как свободные жирные кислоты присутствуют в тканях и плазме крови в небольших количествах и в норме не накапливаются.Синтез триглицеридов происходит из глицерина и жирных кислот (главным образом стеариновой, пальмитиновой и олеиновой). Путь биосинтеза триглицеридов в тканях протекает через образование α-глице-рофосфата (глицерол-3-фосфата) как промежуточного соединения.В почках, а также в стенке кишечника, где активность фермента глицеролкиназы высока, глицерин фосфорилируется за счет АТФ с образованием глицерол-3-фосфата:
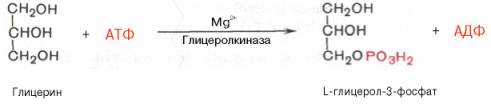
В жировой ткани и мышцах вследствие очень низкой активности глицеролкиназы образование глицерол-3-фосфата в основном связано с процессами гликолиза и гликогенолиза. Известно, что в процессе гли-колитического распада глюкозы образуется дигидроксиацетонфосфат (см. главу 10). Последний в присутствии цитоплазматической глицерол-3-фос-фатдегидрогеназы способен превращаться в глицерол-3-фосфат:

Отмечено, что если содержание глюкозы в жировой ткани понижено (например, при голодании), то образуется лишь незначительное количество глицерол-3-фосфата и освободившиеся в ходе липолиза свободные жирные кислоты не могут быть использованы для ресинтеза триглицеридов, поэтому жирные кислоты покидают жировую ткань. Напротив, активация гликолиза в жировой ткани способствует накоплению в ней триглицеридов, а также входящих в их состав жирных кислот. В печени наблюдаются оба пути образования глицерол-3-фосфата.Образовавшийся тем или иным путем глицерол-3-фосфат последовательно ацилируется двумя молекулами КоА-производного жирной кислоты (т.е. «активными» формами жирной кислоты – ацил-КоА). В результате образуется фосфатидная кислота (фосфатидат):
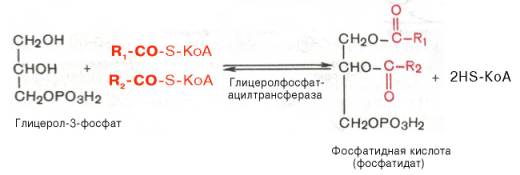
Как отмечалось, ацилирование глицерол-3-фосфата протекает последовательно, т.е. в 2 этапа. Сначала глицерол-3-фосфат-ацилтрансфераза катализирует образование лизофосфатидата (1-ацилглицерол-3-фосфата, а затем 1-ацилглицерол-3-фосфат-ацилтрансфераза катализирует образование фосфатидата (1,2-диацилглицерол-3-фосфата) .Далее фосфатидная кислота гидролизуется фосфатидат-фосфогидро-лазой до 1,2-диглицерида (1,2-диацилглицерола):
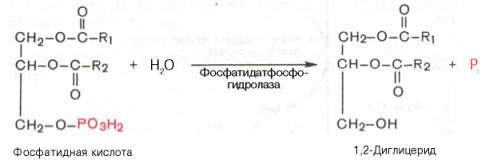
Затем 1,2-диглицерид ацилируется третьей молекулой ацил-КоА и превращается в триглицерид (триацилглицерол). Эта реакция катализируется диацилглицерол-ацилтрансферазой:

Синтез триглицеридов (триацилглицеролов) в тканях происходит с учетом двух путей образования глицерол-3-фосфата и возможности синтеза триглицеридов в стенке тонкой кишки из β-моноглицеридов, поступающих из полости кишечника в больших количествах после расщепления пищевых жиров. На рис. 11.6 представлены глицерофосфатный, дигидроксиацетон-фосфатный и β-моноглицеридный (моноацилглицероловый) пути синтеза триглицеридов.