

Глава II материал и методы исследования
В основу работы положены экспериментальные исследования и клинические наблюдения.
Экспериментальный раздел
Эксперименты поставлены на 48 взрослых беспородных половозрелых собаках обоего пола, массой от 5,9 до 14,1 кг, разделенных на две группы.
В первой группе исследований (24 животных) изучали морфофункциональное состояние ткани кишечника по транскапиллярному обмену, кровенаполнению и биоэнергетике исследуемой ткани, при остром серозном перитоните. Выраженность процессов липопереокнеления определяли по содержанию первичных (диеновые и триеновые коньюгатов и вторичных (маало-новый диальдегпд) продуктов, активность фосфолипазы A2j систему антиок-сидантной защиты оценивали по активности каталазы и супсроксиддисмута-зы в тканях кишки и плазме крови. Качественный и количественный липид-ный состав ткани кишечника и плазмы крови изучали по содержанию ацилг-лицеролов, свободных жирных кислот, эстерифицированного и неэстерифи-цированного холестерола, лизофосфолнлпдов, ефннгомнелина, фосфатидил-холина, фосфатндилсерина, фосфатидилинозита и фосфатидилэтаноламина. Эта группа исследований обозначена как первая (I).
Во второй группе исследований (24 животных) определяли морфофункциональное состояние ткани кишечника, интенсивность процессов перекисного окисления липидов, активность фосфолипазы А2 и ферментов ан-тиоксидантной защиты, качественный и количественный липидный состав ткани кишечника и плазмы крови при остром гнойно-фибринозном перитоните. Эта группа исследований обозначена как вторая (II).
Для получения исходных данных был изучен уровень вышеперечисленных показателей в изучаемых тканях у 10 здоровых животных. Полученные результаты были приняты за норму.
Перитонит моделировали по способу Л.П. Власова (1991) таким образом: Под общим обезболиванием, используя раствор тиопентала-натрия в расчете 0,04 г/кг массы тела, животным в брюшную полость шприцем вводили 20 % каловую взвесь (0,5 мл/кг массы тела животного). В дальнейшем, животным первой группы через одни сутки после введения каловой взвеси выполнили срединную лапаротомию, производили санацию брюшной полости, биопсию серозпо-мышечного слоя кишечника и забор венозной крови. Таким образом, животным моделировали острый серозный перитонит. Во второй группе исследований срединную лапаротомию и санацию брюшной полости с дальнейшим забором венозной крови и биопсией серозно-мышечного слоя кишечника выполняли через двое суток после введения каловой взвеси в брюшную полость. Таким образом, моделировали острый гнойно-фибринозный перитонит. В контрольные сроки (1, 3, 5 суток) после санации брюшной полости животным проводили релапаротомию, биопсию серозно-мышечного слоя кишечника и забор крови из периферической вены для дальнейшего исследования.
В послеоперационном периоде экспериментальным животным проводили антибактериальную (раствор канашщина из расчета 15 мг/кг 2 раза в сутки в/м) и дезннтоксикационную (5% раствор глюкозы и 0,89% раствор хлорида натрия из расчета 50 мл/кг в/в) терапию.
Клинический раздел
Клинический раздел включает 90 больных острым перитонитом.
В первой группе (45 больных) исследовали морфофункциональное состояние кишечника при остром серозном перитоните;
Во второй группе (45 больных) - остром гнойно-фибринозном перитоните.
Подробный анализ больных, подбор их в группы представлен в главе
YI.
В работе применялись следующие методы исследования:
1. Определение окислительно-восстановительного потенциала (ОВП) для изучения электрогенеза тканей. Регистрацию показателя осущест- вляли на универсальном ионометре ЭВ-74 по методике, изложенной В анно- тации к прибору, где рабочий электрод — платиновый (платина 99,99 %, ГОСТ 85888-64), а электрод сравнения - хлорсеребряный.
Контрольными растворами служили 1,0 М феррацианиды составом K4Fe(CN)6/K3Fe(CN)fv Отклонение индикаторной стрелки при Т - 25±1°С составляло 250±4 мВ. Результат выражался в мВ.
2. Определение вено-венозного градиента но методу Ленд пса. Из- менение гистогематической проницаемости регистрировалось по капилляр- нон фильтрации (F) и потере белка (К) на основе следующих расчетов:
100-Hi
F = f00 Цм%
Ht
i
где Ht, - гематокрит венозной крови (контрольные данные); Ht2- гематокриг венозной крови (опытные данные).
100
где Ej- количество белка плазмы (контрольные данные); E^VyPp
Е2- количество белка плазмы (опытные данные); E^V^-Pj,
V, - объем плазмы (контрольные данные);
V2 - объем плазмы (опытные данные);
- процентное содержание белка плазмы (контрольные данные);
Р2 - процентное содержание белка плазмы (опытные данные).
3. Определение коэффициента диффузии кислорода в тканях на основе учета темпа падения диффузного тока восстановления по уравнению И.М. Эпштейна:
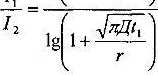
где Ii - сила тока через щ ма; 12 - сила тока через t2, ма; г — радиус электрода, мм. Результат выражался в см /с.
4, Кровенаполнение тканей тонкой кишки. Количество крови в тканях определяли следующим образом: забирали кусочки ткани, который взвешивали и гомогенизировали в дистиллированной воде в соотношении 1:10. Гомогенат ткани центрифугировали в течение 15 минут при 6000 об/мин. Из вены брыжейки участка кишки, ткань которого исследовалась, забирали кровь и стабилизировали 5% раствором цитрата натрия. К 0,1 мл забранной крови добавляли 4,0 мл дистиллированной воды. Гсмолизат центрифугировали при 6000 об/мин в течение 15 минут. В три пробирки вносили по 1,0 мл дистиллированной воды, 1,0 мл реактива и 0,8 мл 3% перекиси водорода. В первую пробирку добавляли 0,2 мл супернатанта ткани, во вторую — 0,2 мл гемолизата (очищенного), а в третью - 0,2 мл дистиллированной воды (контроль на реактивы). Содержимое пробирок перемешивали и центрифугировали в течение 5 минут при 6000 об/мин. Далее измеряли оптическую плотность проб при длине волны 540 им на ФЭКе. Длина оптического пути кювет была 10 мм.
Объем крови в ткани (V, мкл/г) рассчитывали по формуле:
V=((E1 - Е) * 5 * 1000) : ((Е2 - Е) х 2);
где Е1 - экстинция пробы ткани, Е2 - экстинция пробы крови, Е - экстинция пробы контроля на реактивы, 5 — объем крови в пробе (мкл),
2 — масса ткани в пробе (мг),
1000 — коэффициент пересчета из мг в г.
Экстракция липидов из тканей топкой гстпшсн (Хнгпшс Д. А., 1990), Осадок, полученный из гомогенизированных тканей ресуспензировали в стеклянных центрифужных пробирках, содержащих не менее 20 объёмов смеси хлороформ/метанол (2:1 по объёму). Пробирки с экстрактом закрывали стеклянными пробками и оставляли для полной солюбнлизации липидов на 1-1,5 ч. Затем денатурированный белок удаляли центрифугированием, а па-досадочную жидкость сливали в стеклянные центрифужные пробирки. Для полноты экстракции к осадку вновь приливали хлороформ-метанольную смесь и после центрифугирования супернатанты объединяли. К экстракту добавляли одну пятую объема 0,05 М хлорида кальция и тщательно перемешивали. Для разделения на две фазы эмульсию оставляли на ночь в холодильнике или центрифугировали. Верхнюю фазу, состоящую из смеси хлороформ/метанол/вода в соотношении 3:42:47 отбрасывали. Из нижней хлороформной фазы получали препарат суммарных липидов.
Хроматографпческие методы анализа (тонкослойная хроматография), Лштиды фракционировали методом тонкослойной хроматографии на силикагелевых пластинах. Полярные фосфолипиды разделяли на пластинах фирмы Мегк на стеклянной основе, нейтральные липиды фракционировали на силикагелевых пластинах для обращеннофазной тонкослойной хроматографии (Россия). Перед разделением хроматографпческие пластины для удаления связанной воды и повышения разделительной способности силика-геля нагревали в течение 30-60 мин. при температуре 120"С.
Липиды разделяли в прямоугольных хроматографичеекпх камерах, выстланных изнутри фильтровальной бумагой для лучшего насыщения внутреннего объёма парами растворителей. Образцы суммарных препаратов липидов, растворенные в смеси хлороформ-метанол (2:1, по объёму), наносили микрошприцем фирмы "Hamilton" на расстоянии 1 -1,5 см от краев пластины. Для препаративного разделения раствор 2 мг липидов наносили в виде пря
моугольной полосы. При аналитическом разделении нагрузка липидов составляла 100-200 мкг на одну точку.
Фосфолнпиды фракционировали одномерно в системе растворителей хлороформ+метанол+ледяная уксусная кислота+вода (60:50:1:4, по объёму).
Нейтральные липиды разделяли, элюируя хроматографпческие сплика-гелевыс пластины в системе растворителей гексан+диэтнловый эфир +уксусная кислота (90:10:1, по объёму). На хроматограммах липиды обнаруживали 10% раствором фосфорномолибденовой кислоты в этаноле, после нагревания в течение 10 минут при температуре 90 С проявлялись интенсивно-синие пятна липидов.
Лнпиды на хроматограммах идентифицировал н с помощью специфических окрашивающих реагентов и свидетелей. Фосфолнпиды выявляли с помощью универсального реагента (Vaskovsky V.E. et al., 1975), приготовляемого из исходного реагента. Исходный реагент готовили, добавляя к 10 г натрия молибденовокислого в 60 мл 4 н НС1 0,4 г солянокислого гидразина в 14 мл 4 н НС1 и, нагревая смесь на водяной бане в течение 20 мин. Затем охлаждали, добавляли 14 мл концентрированной серной кислоты и доводили объем до 100 мл дистиллированной водой. Для опрыскивания хроматографи-ческих пластин готовили смесь, состоящую из одного объема исходного реагента и семи объемов 7 н серной кислоты.
На хроматограммах после обработки указанным реагентом фосфолнпиды обнаруживались в виде сине-голубых пятен. Фосфатидилэтаноламнн и фосфатидилсерин обнаруживали нингидршювым реактивом, опрыскивая хроматографпческие пластинки 0,15% раствором нингидрина в ацетоне и, нагреваядо 100ВС
Аминосодержащие фосфолнпиды окрашиваются в краснофиолетовый цвет. Холинсодержащие фосфолнпиды обнаруживали с помощью реагента Драгендорфа (Marinetti Р., 1976). Реагент Драгендорфа готовили перед употреблением. Смешивая 20 мл раствора I, состоящего из 1,72 г основного висмута азотнокислого в 100 мл 20% ледяной уксусной кислоты и 5 мл раствора
1% приготовленного из 10 г йодистого калия растворенных в 25 мл воды. После опрыскивания холннсодсржащие фосфолнпиды окрашивались в оранжевый цвет.
Количественное определение липидов. Количественное определение липидов проводили непосредственно на хроматограммах депептометри-ческим методом после их проявления 5% фосфорнованилиновоп кислотой в этаноле. Молекулярный анализ проводили на денситометре Model CS-670 (BIO-RAD, США) с соответствующим программным обеспечением (Phosphor Analyst/PS Sowlware).
Определение диеновых п трненоных коныогатов. Спектрофото-мстрический метод определения продуктов ПОЛ основан на том, что диеновые коныогаты обладают характерным поглощением при длине волны 232233 нм [Ганстон, 1986]. Липиды из тканей и плазмы крови экстрагировали хлороформ-мстанольной смесью. Суммарный препарат липидов высушивали досуха на роторном вакуумном испарителе и остаток липидов растворяли в гексапе. Спектр поглощения регистрировали при длинах волн 190-275 нм на спектрофотометре СФ-46 (Россия).
Окпсленность липидов оценивали по величинам индексов окисленно-сти, рассчитываемых на 1 мг липидов по определению отношения А 232 /А 215 и А 275/А 215 (А - оптическая плотность при указанных длинах волн). Содержание диеновых и триеновых коныогатов выражали в усл.ед./мг липидов.
Определение содержания малонового дпальдегнда. Содержание малонового диальдегида оценивали в реакции с 2-тиобарбитуровой кислотой (ТЕК). Для определения антиокислительной активности липидов предварительно проводили индукцию липопереокисления раствором сульфата железа в концентрации 5 мкмоль в течение часа. Для этого к 1 мл плазмы крови или тканевого гомогената добавляли 3 мл 1% фосфорной кислоты, содержащей 0,5 ммоль ЭДТА и 1 мл 0,5% раствора ТЕК. Образцы перемешивали и инкубировали 45 мин при 100°С. Затем образцы охлаждали и приливали 4 мл н
бутанола, тщательно встряхивали и центрифугировали 15 мин при 1500 g. В верхней бутанольной фазе регистрировали спектр поглощения в области 515550 нм. Определяли оптическую плотность при 532 нм, используя в качестве базовых точки спектра при 515 и 550 нм. Содержание ТБК-реагирующих продуктов выражали в имоль/г белка.
10. Определение активности фосфолипазы А2, Солюбилизацию мембранных белков тканей и плазмы крови проводили с использованием 1,0% раствора тритон Х-100. Для этого к навескам ткани и плазме крови прибавляли 0,5 мл 10 мМ раствора трис-НСЬ-буфера с 1% тритон Х-100 (15 мМ), рН 8,0. Гомогенизацию тканей, с целью полного извлечения белков, проводили с помощью стеклянного гомогенизатора со шлифованным пестиком (при температуре 37 С в течение 10 минут).
Для ннгибировапия сернновых протеаз в гомогенаты добавляли фенил метансульфонил фторид (10 мкМ). Для определения активности фермента к 1 мл ферментного препарата прибавляли 1 мл 10 мМ трис-НСЬ-буфера (рН 8,0), содержащего 150 мМ тритон Х-100, 10 мМ СаС12 и субстрат (1,2 мМ). В качестве субстрата использовали фосфатндилхолипы яичного желтка. К 1 мл тканевого гомогената яичного желтка приливали 3,75 мл емсен хлороформ-метанол (1:2, по объёму). Содержимое пробирки перемешивали и выдерживали в холодильнике в течение 2-3 часов, периодически встряхивая, затем центрифугировали 20 мин при 3000 об/мин. Супернатант переносили в другую пробирку, а к осадку приливали 4,75 мл смеси, состоящей из одной части хлороформа, двух частей метанола и 0,8 части воды. После повторной экстракции содержимое пробирки разделяли центрифугированием и суперна-танты объединяли. Для промывки и разделения на две фазы добавляли 2,5 мл хлороформа и 2,5 мл воды. Образующуюся эмульсию расслаивали центрифугированием в течение 30 мин при 3000 об/мин. Нижний хлороформный слон упаривали досуха с помощью роторного испарителя или в токе азота. Сухой остаток липидов растворяли в смеси хлороформ-метапод (2:1, по объёму) и
использовали в качестве субстрата (лецитин яичного желтка) для определения активности фосфолипазы А2.
Регистрацию каталитической деятельности фермента проводили на установке, состоящей из иономера ЭВ-74, электродной системы (электрод сравнения ЭВЛ-IM, измерительный электрод ЭСЛ-43-07), ультра-термостата и микробюреткн.
Активность Фосфолипазы А2 оценивали титрометрнческим методом с помощью нейтрализации 0,02 М NaOH карбоксильных групп, выделяющихся свободных жирных кислот. Расчет проводили по калибровочной кривой, построенной по пальмитиновой кислоте и выражали в мкмоль/с/г белка.
11. Определение активности каталазы. Фотометрический метод оп- ределения активности каталазы основан на способности перекисей образовы- вать с молнбдатом аммония стойкий окрашенный комплекс. К 0,1 мл плазмы крови или эритроцитов приливали 2 мл 0,03% перекиси. В холостую пробу вместо сыворотки добавляли 0,1 мл дистиллированной воды. Реакцию оста- навливали через 10 мин добавлением 1 мл 4% раствора молибдата аммония. Образцы центрифугировали при 8000 об/мин в течение 40 мин.
Оптическую плотность измеряли на спектрофотометре при длине 410 нм. Активность каталазы выражали в мг ЬЬО/мин/г белка.
12, Определение активности супсроксиддисмутазы- К 500 мг ткани приливали 3 мл фосфатного буфера (рН 7.4) и гомогенизировали до появле- ния однородной суспензии. После центрифугирования при 5000 об/мин в те- чение 15 минут отбирали 0,5 мл супернаганта и вносили в центрифужную пробирку, содержащую 1 мл хлороформно-спнртовой смеси (2:1, по объёму). Полученную смесь охлаждали, тщательно перемешивали и для удаления ге- моглобина центрифугировали. Водно-спиртовой слой, содержащий фермент отбирали и добавляли несколько капель насыщенного раствора КН2РО4. Ферментный препарат получали разбавлением полученной фазы в 20 раз.
В состав реакционной среды входили питросииий тетразолий (57 мкмоль), НАДН (98,5 мкмоль), феназннметасульфат (16 мшоль) и 0,2 мл
ферментного препарата, Нитросиний тетразолий используется как индикатор, способный акцептировать электроны и восстанавливаться до формазана, имеющего максимум поглощения при 560 нм. Восстановленная форма би-формазан окрашена от синего до черного цвета. Нитросиний тетразолий быстро восстанавливают в присутствии феназинметасульфата флавопротеино-вые ферменты.
Феназинметасульфат используется в реакциях определения активности ферментов как переносчик электронов между ферментами и молекулярным кислородом или солями тетразолия. Реакция протекала 10 минут в 0,5 моль фосфатном буфере (рН 8,3) с ЭДТА (0,1 ммоль) при 25° С в аэробных условиях. В контрольную пробу ферментный препарат не вносили. Активность СОД рассчитывали по формуле:
А = Т%/(100%-Т%),
где А - активность фермента в условных единицах, рассчитанная на 1 мг белка,
Т % - процент торможения реакции восстановления нитроси-
него тетразолия в пробе за минуту. Полученные цифровые данные обрабатывали методом вариационной статистики, с использованием критерия t Стьюдента. Вычисления производили на CRU 199 MHz "Pentium-MMX" с помощью программы Microsoft Excel 2000. Использован текстовый процессор Microsoft Word 2000. Динамика показателей отражена на графиках, построенных с помощью программы электронных таблиц Microsoft Excel 2000.