

Техника безопасности при работе с культурами клеток
Правила техники безопасности в лаборатории, где работают с культурами клеток можно разделить на две основные группы:
Общие правила техники безопасности ( электричество, газовые баллоны, работа с жидким азотом, вентилируемость помещений и т.д.)
Правила биологической безопасности.
Правила экологической безопасности.
Общие правила техники безопасности.
В целях противопожарной безопасности с учетом максимальной изолированности от окружающей среды культуральные боксы должны быть обеспечены средствами пожаротушения
Аппаратура и оборудование должны размещаться в лаборатории таким образом, чтобы обеспечивалось наибольшее удобство в работе и наименьшие затраты времени на переходы.
Запрещается включать ток, если в лаборатории пахнет газом. Предварительно необходимо определить и ликвидировать утечку газа и проветрить помещение. Место утечки газа определяется с помощью мыльной воды. Все мероприятия по устранению утечки газа должны проводиться аварийной службой Гор.газа.
Запрещается пробовать на вкус и вдыхать неизвестные вещества.
Запрещается в боксах и комнатах, предназначенных для работы с клеточным материалом, курить, хранить и принимать пищу, выращивать цветы в вазонах.
Запрещается работать без специальной одежды и предохранительных приспособлений;
Запрещается выполнять работы, не связанные с заданием.
В целях исключения поражений электрическим током запрещается переносить включенные приборы и ремонтировать оборудование, находящееся под током.
При эксплуатации приборов и аппаратов необходимо строго руководствоваться правилами, изложенными в техническом паспорте. Приборы должны быть заземлены, если этого требует инструкция по их эксплуатации. Ежемесячно проверяется исправность электроприборов. Особое внимание уделяется круглосуточно работающим электроприборам – углекислотному инкубатору, например. При нарушении работы электроприбора (запах, выделение дыма, изменение характера шума и т.д.) прибор отключают от сети и не используют до проверки и проведения необходимого ремонта.
Во время измерения давления углекислого газа или кислорода в баллонах для инкубатора, нужно применять манометры, предназначенные только для этих газов.
При работе с газами в баллонах под давлением, запрещается хранить баллоны в рабочем помещении, переносить большие баллоны на руках, выпускать газ без требуемой регулировки и проверки соединений баллона с установкой, использовать немеченные баллоны.
Запрещается проводить работы с жидким азотом без защитных средств рук и глаз, а также в плохо вентилируемом помещении.
Правила биологической безопасности. Эти правила обусловлены главным образом тем, что до сих пор до конца не ясно с чем на самом деле исследователь имеет дело, какой чистоты материал он использует (микоплазмы, вирусы), какой потенциал несет клетка в культуре, как меняется этот потенциал в ходе эксперимента.
Необходимо крайне осторожно обращаться с колюще-режущими предметами, особенно непосредственно вступающими в контакт с культурой клеток.
Необходимо избегать контакта прлитых культуральных сред, ее компонентов и других веществ неорганической и органической природы с открытыми участками тела.
Необходимо знать свою индивидуальную чувствительность к используемым в лаборатории веществам и их парам (дезинфектантам и т.д. ).
Необходимо регулярно проводить дезинфекцию внутренних камер ламинара и инкубатора дезинфектантами и УФ-облучением.
Необходимо регулярно до и после проведения работ проводить дезинфекцию помещения влажной уборкой и УФ-излучением.
Необходимо до и после работы дезинфецировать сменную одежду.
Правила экологической безопасности.
Отработанный биоматериал перед утилизацией (животные-доноры, ткани, культуры клеток) необходимо поместить в целлофановый пакет, залить дезинфицирующим раствором и герметично закрыть.
Перед утилизацией культуральный пластик одноразового использования должен быть обработан дезинфицирующим раствором. Отработанные иглы шприцов должны быть кроме того упакованы перед утилизацией так, чтобы о них нельзя было случайно пораниться.
Работа № 2.
Введение изолированных клеток в культуру. Получение первичной культуры фибробластов легкого и роговицы.
Цель работы: получить первичную культуру фибробластов легкого и роговицы крысы (возраст 2 недели) методом трипсинизации ткани и методом естественной миграции клеток на субстрат. В ходе работы овладеть навыками правильной работы с культуральной посудой и расходными материалами, реактивами.
Протокол работы №1. Получение первичной культуры методом трипсинизации ткани.
! |
|
1. Подготовить рабочее место.
2. Усыпить животное диэтиловым эфиром.
3. Под ламинаром декапитировать животное, смочить грудную клетку и область глазницы этиловым спиртом, произвести вскрытие грудной клетки и забор биоптата легкого (2х1 см) и глазных яблок. При выполнении операции следует избегать попадание крови на забираемые ткани. В случае необходимости - промыть ткани от крови в стерильном буферном растворе Дюльбекко.
4. Одно глазное яблоко и фрагмент легкого размером 5х5 мм поместить в отдельные пробирки с культуральной средой Дюльбекко. Поместить в термостат на 37 0С. Эти фрагменты ткани будут использоваться для отработки второго метода выделения первичной культуры.
5. Остаток тканей поместить пинцетом ткани в отдельные конические пробирки с 3 мл 0.1% раствором трипсина, нагретого до 370С. Используя ножницы мелко измельчить ткани. Закрыть пробирки крышкой и поставить на инкубацию в термостат при 370С на 15 минут.
5. Отфильтровать через нейлоновую сетку с диаметром пор 20-25 мкм (диаметр фибробластов в суспензии составляет около 10-15 мкм).
6. Отцентрифугировать фильтрат при 500 об.мин в течении 5 минут. При этом клетки и мелкие фрагменты ткани уходят в осадок.
7. Отобрать пипеткой осадок и произвести посев клеток в приготовленные чашки с культуральной средой. Закрыть чашки.
8. Поместить чашки в углекислотный инкубатор (37 0С, 10% СО2).
9. Через 24 часа пронаблюдать за изменением морфологии клеток при их прикреплении и распластывании по дну чашки. Отделить мелкие фрагменты ткани и неприкрепившиеся клетки заменой питательной среды.
10. В течении недели каждые 24 часа наблюдать за увеличением количества клеток и образованием монослоя. Производить замену питательной среды каждые 48 часов.
|
1 – клетки в суспензии.
2-3 – распластывание клеток по подложке и рост культуры.
|
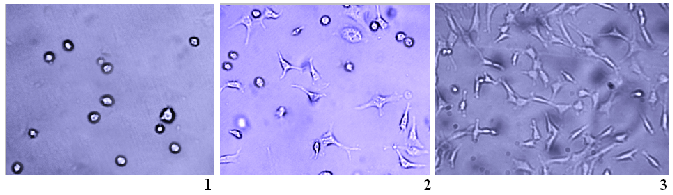
Протокол работы №2. Получение первичной культуры методом естественной миграции клеток из ткани на субстрат.
! |
|
1.Пробирки с фрагментами легкого и глазное яблоко (отобранные в 4-м пункте Протокола №1 для второй части работы) достать из термостата и поместить под ламинар.
2.Захватить ткань глазным пинцетом и отрезать как можно меньший кусочек ткани.
3.Снять отрезанный кусочек с ножниц иглой и «посадить» его на дно пустой культуральной чашки. Рядом поместить еще несколько кусочков.
4. Используя пинцет, накрыть кусочки покровным стеклом, слегка его придавив пинцетом ко дну чашки, слегка расплющивая, но не давя фрагменты ткани.
5. Заполнить чашки культуральной средой так, чтобы стекла были слегка покрыты средой.
6. Закрыть чашки и поместить их в углекислотный инкубатор (37 0С, 10% СО2).
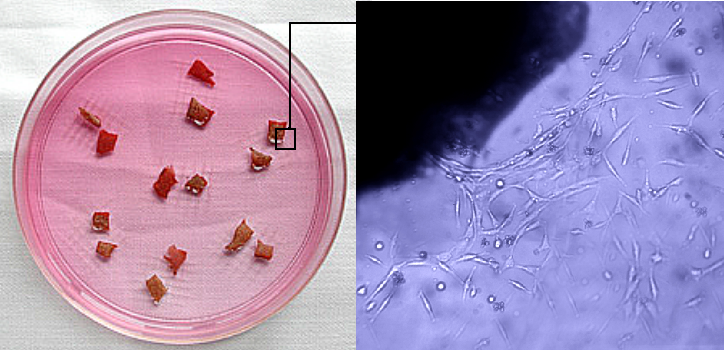
7. Через неделю начать наблюдать за кусочками каждые 24 часа. К этому времени клетки начинают мигрировать из ткани на субстрат, вокруг кусочков образуется так называемая зона (или граница) роста. Вначале появляются единичные фибробласты, которые заметны в виде длинных веретеновидных клеток. При образовании зоны роста необходимо произвести замену среды.
8. Когда пространство между кусочками будет заполнено клетками, покровное стекло снимают, убирают кусочки ткани, производят замену среды. В течении следующей недели каждые 24 часа наблюдают за образованием монослоя клеток.
Миграция клеток на субстрат из фрагмента ткани
|
Почему клетки в суспензии имеют сферическую форму?
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
За счет чего происходит прикрепление клетки к подложке (субстрату) и какие основные механизмы миграции клетки из ткани на субстрат и движения клетки по субстрату?
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Работа № 3. Субкультивирование. Определение жизнеспособности клеток.
Цель работы: Провести пересев клеток первичной культуры фибробластов, полученных в работе №2. Построить кривую роста культуры.
! |
|
Протокол работы.
1. Снять клетки с дна культуральной чашки. Для этого заменить весь объем питательной среды из культуральной чашки с монослоем первичной культуры фибробластов на 2-3 мл подогретого до 370С 0.25% раствора трипсина. Помещают чашки в инкубатор.
2. Инкубируют в термостате 15 минут при 370С. Диссоциацию клеток и их открепление от дна чашки можно контролировать, просматривая клетки под микроскопом.
3. По окончании трипсинизации, слегка наклоняют чашку (чтобы жидкость не касалась края чашки!) и пипеткой отбирают получившуюся суспензию клеток в пробирку.
4. Центрифугируют при 500 об.мин 5 минут. Супернатант удаляют, а осадок клеток несколько раз отмывают от раствора трипсина 2 мл культуральной средой Дюльбекко (370С).
5. После отмывки, клетки ресуспендируют в 2 мл культуральной среды.
6. Оценивают количество живых клеток методом прижизненного окрашивания трипановым синим. Для этого из полученной в пункте 5 суспензии клеток, берут аликвоту объемом 10 мкл и добавляют 90 мкл 0,1% раствор трипанового синего. Подготовка гемоцитометра. Шлифованное покровное стекло притирают к предметному стеклу гемоцитометра до появления колец Ньютона, так, чтобы покрыть заштрихованные области. Это приводит к образованию камеры с фиксированным объемом, поскольку края предметного стекла подняты над заштрихованной поверхностью ровно на 0,1 мм. Каждая заштрихованная область состоит из 9 больших квадратов размером 1х1мм, т. е. объем, ограниченный каждым большим квадратом, оказывается равным 1мм х 1мм х 0,1 мм = 0,1 мм3.
Заполняют счетную камеру гемоцитометра окрашенной суспензией клеток за счет капиллярного всасывания, не переполняя каналы камер. Оставляют на несколько минут постоять, чтобы клеткы осели и приняли стабильное положение.
|
Живые клетки не окрашиваются трипановым синим, мертвые – окрашиваются.
|
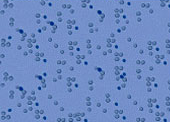
Подсчет окрашенных (мертвых) клеток. Используется объектив x10. Клетки подсчитываются в 5 больших квадратах 1х1 мм в направлении слева направо сверху вниз (начинают с левого верхнего квадрата, заканчивают средним). Клетки на границе квадратов подсчитывают по правилу – считать клетки только на верхней и левой границах. Клетки подсчитываются в обеих камерах (верхней и нижней). В каждой камере 9 квадратов, подсчитывают в 5 из них, в верхней и нижней камерах. Общее количество квадратов – 10.
Среднее количество клеток на квадрат = общее количество клеток в 10 квадратах
количество квадратов (10).
К-во клеток в 1 мл. суспензии = среднее число клеток в квадрате (1x1 мм.) × фактор дилюции × 10 000.
К-во клеток в суспензии = к-во клеток в 1 мл. × общий объём суспензии (в мл.).
|
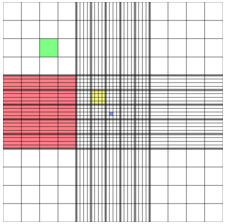
Решетка цитометра:
Синий квадрат – 0,0025 мм2, 0,25 нл. Желтый квадрат – 0,04 мм2, 4 нл. Зеленый квадрат –0,0625 мм2, 6,25 нл. Красный квадрат - 1 мм2, 100 нл. |
Выразить количество живых и мертвых клеток в % от общего количества.
7. Суспензию клеток, полученную в пункте 5 переносят в культуральный флакон с вентилируемой крышкой. В флакон предварительно наливают культуральную среду, слоем не более 5 мм. Суспензию клеток стараются распределять по флакону равномерно.
8. Помещают флакон в инкубатор (37 0С, 10% СО2). Флакон с пересаженными растущими фибробластами просматривают ежесуточно под микроскопом. Через 24-48 часов производят подсчет распластавшихся клеток и замену культуральной среды. Далее каждые 24 часа подсчитывают общее количество клеток в культуре. Обычно через 4-7 суток образуется монослой клеток.
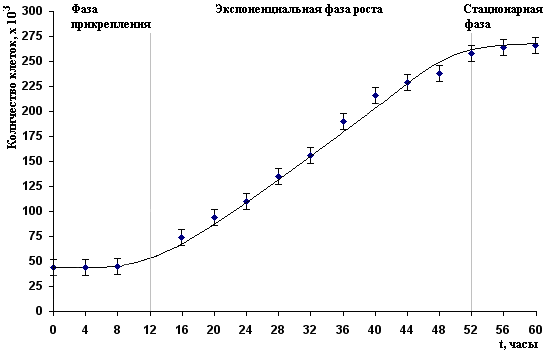
9. На основании полученных данных строят зависимость количества клеток в культуре от времени культивирования.
В чем принцип метода выявления живых и мертвых клеток при помощи трипанового синего?
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Опишите особенности поведения и метаболической активности клеток в трех фазах кривой роста культуры. Будут ли различаться (если да, то в чем различия) кривые роста нормальных и раковых клеток?
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Работа № 4. Криоконсервация клеток. Введение клеток в культуру после заморозки.
Цель работы: Произвести криоконсервацию фибробластов с последующей разморозкой и оценкой их жизнеспособности.
! |
|
Протокол работы №1.Заморозка клеток.
1.Клетки, полученные в работе №3, снять с дна культуральных флаконов трипсинизацией и отмыть от трипсина, как описано в пунктах 1-4 предыдущей работы.
2.К осадку клеток добавить 1 мл среды Дюльбекко, содержащей 25% фетальной сыворотки (25%FBS-среда). Ресуспендировать клетки.
3. Отобрать аликвоту суспензии клеток и произвести в гемоцитометре подсчет общего количества клеток в без какого либо окрашивания. Рекомендуется для заморозки брать не менее 0.5-1 млн клеток.
4.Отдельно приготовить раствор-криопротектор: 800 мкл 25%FBS-среды+200 мкл 10% ДМСО (в 25%FBS-среде). ДМСО (DMSO, Диметилсульфооксид) используется как криопротектор.
5. К 1мл суспензии клеток в среде добавить 1 мл раствора-криопротектора.
6. Поместить смесь в предварительно охлажденные до +40С криопробирки.
7. Криопробирки поместить в камеру замораживателя. Придерживаться режима охлаждения 2 0С в минуту. По достижении температуры -700С, контейнер с пробирками перемещается в сосуд Дюара с жидким азотом.
Протокол работы №2. Размораживание клеток.
Примечание: замороженные клетки достаточно хрупкие (плохо переносят пипетирование) и требуют аккуратного обращения. Клетки размораживают быстро и помещают сразу в полную ростовую среду. Если клетки чувствительны к криоконсервантам, их отмывают через центрифугирование, чтобы удалить криоконсервант, и затем делают посев. Так, например, при работе с клетками человека отмывку делают 3 раза.
! |
|
1.Криопробирками достать из азота и выдержать на воздухе 15 минут.
2.Поместить криопробирки на водяную баню (37 0С) до остаточного льда в пробирке 10%. Переместить пробирки в ламинар.
3. Оттаявшую суспензию клеток аккуратно ресуспендировать в 10 мл культуральной среды ( 37 0С) с последующим центрифугированием 500 об.мин (80 g, 2-3 мин).
4.Полученный осадок клеток ресуспендировать в 2-3 мл среды.
5. Произвести подсчет общего количества, а также живых и мертвых клеток. Выразить жизнеспособность в %.
6. Произвести посев клеток в культуральные флаконы.
Какой механизм криопротекторного действия ДМСО? __________________________________________________________________________________________________________________________________________________________
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Почему заморозку и разморозку клеток следует производить со скоростью 1-2 град. цельсия в минуту?
_________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Почему после размораживания клеток их необходимо отмыть от ДМСО?
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Работа № 5. Окрашивание ядер и лизосом нормальных и апоптических фибробластов акридиновым оранжевым.
Цели работы: Оценить жизнеспособность фибробластов, растущих в атмосфере с 10% CО2 и среде без углекислого газа методом окрашивания их ядер и лизосом акридиновым оранжевым.
Протокол работы №1. Окраска ядер фиксированных клеток акридиновым оранжевым.
Примечание: Для нормального функционирования фибробластов необходимо наличие СО2 в атмосфере культивирования. Культивирование клеток в отсутствии углекислого газа в среде приводит к замедлению роста культуры, увеличению апоптических клеток в культуре и ее гибели. Акридиновый оранжевый – флуорохроматический краситель, который связывается с нуклеиновыми кислотами клеток. Под действием света длиной волны 430 нм в нормальных клетках ядра окрашиваются в зелёный цвет, а в апоптотических в оранжево-красный. Принцип такой дифференциальной окраски - молекулы акридинового оранжевого по-разному взаимодействуют с однонитевыми и двунитевыми молекулами РНК и ДНК. Окрашенные двунитевые молекулы нуклеиновых кислот флуоресцируют зеленым светом, а однонитевые – красным. В некоторых апоптоических клетках, даже можно увидеть фрагментацию ядра. Перед покраской образцы необходимо обработать РНКазой (РНК также окрашивается этим красителем в оранжевый-красный цвет).
! |
|
1. Для работы предварительно произвести посев вторичной культуры фибробластов в культуральные чашки и поместить в СО2-инкубатор с 10% углекислого газа. Через 3-4 суток произвести замену культуральной среды, а одной и групп отключить подачу СО2. Через 4 суток инкубации в различных условиях производят окрашивание ядер клеток и оценку их состояния в культуре.
2. Приготовить концентрированный раствор акридинового оранжевого: 1 мг красителя на 1 мл бидистиллированной воды (может храниться в темноте при 4оС).
3. Из культуральных чашек с клетками удалить культуральную среду, и залить свежеприготовленным 4% раствор параформальдегидом (в 1х PBS pH 7,0 ). Инкубировать при комнатной температуре в течение 15 мин.
4. Удалить параформальдегид и промыть два раза (выдержка 5 мин) ацетатным буфером (pH 4,2).
5. Залить фиксированные клетки на 15 мин свежеприготовленным рабочим раствором акридинового оранжевого (1 часть концентрированного раствора на 9 частей ацетатного буфера (pH 4,2)).
6. Удалить краситель и промыть в 2 сменах ацетатного буфера (pH 4,2) по 2 мин. Дать высохнуть препаратам в потоке воздуха комнатной температуры без доступа света.
7. Произвести флуоресцентное микроскопирование препаратов. Условия флуоресценции: длина волны возбуждения – 430 нм, отсекающий фильтр- 515 нм.
Оценить морфологию ядер клеток растущих в условиях нормальной для их роста атмосферы (10% CO2) и в отсутствии углекислого газа.
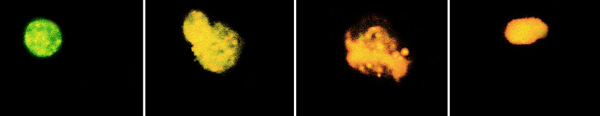
Нормальная клетка Ранний апоптоз Поздний апоптоз Некроз
Протокол работы №2. Прижизненная окраска лизосом нормальных и апоптических клеток акридиновым оранжевым.
! |
|
1. Для работы предварительно произвести посев вторичной культуры фибробластов в культуральные чашки и поместить в СО2-инкубатор с 10% углекислого газа. Через 3-4 суток произвести замену культуральной среды, а одной и групп отключить подачу СО2. Через 4 суток инкубации в различных условиях производят окрашивание лизосом клеток и оценку их состояния в культуре.
1. Приготовить концентрированный раствор акридинового оранжевого – 1 мг красителя в 2 мл буфера Макильвейна (McIlwaine’s buffer), pH 7,3.
Приготовление буфера Макильвейна: смешать 18,17 мл 0,2 М Na2HPO4 и 1,83 мл лимонной кислоты (конечный объем 20 мл). Полученный буферный раствор должен иметь рН 7,4. Довести рН буфера до 7,3 минимальным количеством HCl. Профильтровать полученный раствор через шприцевой фильтр с диаметром пор 0,2 мкм.
2. Приготовить рабочий раствор акридинового оранжевого – 0,1 мл концентрированного раствора развести в 9,9 мл буфера.
3. Промыть клетки от культуральной среды PBS-буфером Дюльбекко.
4. Инкубировать клетки 5 мин в свежеприготовленном рабочем растворе красителя (370С, 10 % СО2).
5. Отмыть клетки от красителя (пару раз по несколько секунд) буфером Макильвейна, заключить препарат в том же буфере и исследовать под микроскопом. Лизосомы живых клеток флуоресцируют оранжевым цветом (А). Мертвые клетки имеют ярко-красную флуоресценцию цитоплазмы и ярко-зеленую ядер (Б).
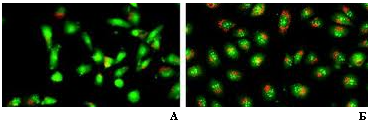
В чем принцип дифференциальной окраски ДНК акридиновым оранжевым? Завсисит ли спектр флуоресценции этого красителя от pH (если да, то как)? Какие еще ДНК-специфичные флуоресцентные красители существуют?
_______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Работа № 7. Иммунохимическое изучение структуры цитоскелета клеток.
Цель работы: Произвести окрашивание флуоресцентными антителами к актину актиновых фибрилл цитоскелета фибробластов с нативным (нормальным) и фрагментированным цитоскелетом.
Примечание: моноклональные антитела связанные (коньюгированные) с каким либо флуоресцентным красителем позволяют специфически окрашивать тот или иной структурный элемент клетки. Так FITC-коньюгированные антитела к F-актину (где FITC- флюоросцеин изотиоционат, краситель) позволяют визуализировать актиновые фибриллы в клетке, их ориентацию, локализацию и т.д.
Цитохалазин В – модификатор цитоскелета, препятствует образованию в клетках актиновых фибрилл, вследствии чего клетки частично или полностью теряют возможность двигаться по субстрату.
Протокол работы:
! |
|
1. Для работы заранее произвести посев клеток вторичной культуры в две культуральные чашки. На 5-е сутки роста культуры оценивают количество и морфологию клеток, особое внимание обращая на фибробласты с морфологией, свидетельствующей об их движении.
2. За сутки до начала окрашивания (4-е сутки культивирования) в одну из чашек внести цитохалазин В (10 мкг на мл среды) и инкубировать 24 часа (370С, 10% СО2).
3. Снять чашки с инкубации. Удалить культуральную среду, и залить свежеприготовленным 4% раствор параформальдегидом (в 1х PBS pH 7,0 ). Инкубировать при комнатной температуре в течение 15 мин.
4. Удалить р-р параформальдегида из чашки и промыть клетки 1х PBS 2-а раза по 2-3 минуты.
5. Смочить препарат клеток раствором БСА-Tween содержащим 3% БСА (бычий сывороточный альбумин) и 0,02% Tween 20 в 1х PBS. Закрыть герметично чашки и инкубировать 30 минут при 37оС и 95% влажности.
6. Удалить раствор БСА- Tween из чашек и смочить препарат клеток раствором антител (разведенных в растворе БСА- Tween). Закрыть герметично чашки и инкубировать 30 минут при 37оС и 95% влажности.
7. Произвести отмывку препарата от раствора антител в 3 сменах 0,02% Tween 20 на 1х PBS по 5 мин при 37оС плавно покачивая чашки.
8. Заключить препарат в фотозащитную среду (5% N-пропилгаллат и 95% нефлуоресцирующего глицерина ) и исследовать под микроскопом соблюдая условия флуоресценции: длина волны возбуждения 480 нм, дихроичное зеркало 505 нм. Эмиссия (спектр свечения) коньюгированных с этим красителем антител сосавляет 535 нм (зеленый цвет).
9. Оценить морфологию клеток нормальных, и культивированных с цитохалазином, изменение локализации актина в клетках обеих групп.
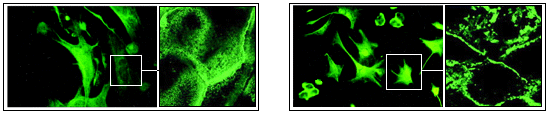
Нормальные фибробласты После инкубации с цитохалазином
Какие флуоресцентные красители кроме FITC могут быть конъюгированы с антителами?
Механизм действия цитохалазина на цитоскелет клетки. Какие модификаторы цитоскелета кроме цитохалазина еще существуют?
________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Работа № 8. Выделение геномной ДНК из культуры клеток.
Введение.
Выделение ДНК из культуры клеток эукариот является первым этапом работ, связанных с проведением многих современных методов молекулярной биохимии – полимеразной цепной реакции, рестрикции, гибридизации и др..
Традиционные методы выделения с помощью фенола и хлороформа не подходят для культур клеток, т.к. эти компоненты могут реагировать с культуральным пластиком и не обеспечивают необходимую полноту и чистоту выделения.
В настоящее время существует ряд наборов для выделения как ДНК так и РНК из культур бактериальных и животных клеток, тканей, бактерий, крови, и др.
Современные требования к методу выделения ДНК\РНК из культур животных клеток:
Полное экстрагирование нуклеиновых кислот из клеток.
Минимизация потерь и фрагментации при очистке.
Как можно более высокая степень очистки.
Не использование токсичных реактивов.
Процесс выделения ДНК\РНК из пробы должен исключать перекрестную контаминацию (загрязнение пробы) и требует строгой асептики (средств индивидуальной защиты, одноразовых наконечников и пробирок, отдельных автоматических пипеток, ультрафиолетовое облучение рабочего места и всей лаборатории), а также предусматривает оптимальную планировку помещения лаборатории.
В работе используются фибробласты легкого крысы, находящиеся в стационарной фазе роста (монослой) в количестве не менее 5×106.
Для выделения гномной ДНК используется кит-набор для выделения нуклеиновых кислот из биологического материала «ФБиоНуклео» (ООО “Фрактал Био”, Россия). Принцип действия заключается в селективной сорбции нуклеиновых кислот на силикатной мембране внутри микроцентрифужных колонок-пробирок (spin columns) в растворах с высокой концентрацией хаотропной соли, с последующей элюцией ДНК.
Задача.
Выделить геномную ДНК из культуры фибробластов кожи крысы методом сорбции ДНК.
Протокол работы.
Внимательно ознакомтесь с инструкцией к набору и техникой безопасности.
Состав набора:
Лизирующий буфер (на основе гуанидинтиоцианата).
Промывочный буфер
Буфер для элюции.
Микроцентрифужные пробирки с вкладышем-колонкой.
Собирающие пробирки.
! |
|
Дважды промыть монослой клеток стерильным фосфатным буфером Дюльбекко (DPBS).
Соскрести клетки скребком, суспендируя в минимальном объеме, необходимом для центрифугирования DPBS.
Центрифугировать при 1000 об\мин 5 минут для получения осадка клеток.
Отобрать в пробирку типа «Эпендорфф» 50 мкл клеток.
Добавить 100 мл воды культурального качества, тестированной на отсутствие ДНК-аз
К 100 мкл пробы добавить 800 мкл лизирующего буфера.
Параллельно береться проба на отрицательный контроль – 100 мкл воды.
Инкубировать микропробирку со смесью 5 минут при комнатной температуре, периодически встряхивая на вортексе.
Центрифугировать при 13000 об\мин при комнатной температуре 5 минут для осаждения клеточной мембраны.
Осторожно, не взбалтывая осадок (!) перенести 800 мкл супернатанта на прилагаемую к набору колонку-вкладыш в центрифужной пробирке. Центрифугировать при 13000 об\мин при комнатной температуре 1 минуту.
Удалить сток, вытащив вкладыш-колонку из центрифужной микропробирки.
Поместить колонку назад и нанести на нее 450 мкл промывочного буфера. Центрифугировать при 13000 об\мин при комнатной температуре 1 минуту. Операцию промывки повторить трижды.
Перенести вкладыш-колонку в собирательную пробирку. Не касаясь наконечником дозатора колонки (!) нанести в ее центр 50 мкл промывочного буфера. Центрифугировать при 13000 об\мин при комнатной температуре 1 минуту. Собрать содержащий ДНК элюат.
Померять спектрофотометрически абсорбцию выделенной ДНК при двух длинах волн – 260 и 280 нм. Рассчитать соотношение А260/А280. Значение соотношения А260/А280 в пределах 1,8-1,9 свидетельствует о высоком уровне чистоты выделенной ДНК.
Работа № 9. Разделение молекул ДНК по молекулярной массе методом электрофореза.
Введение.
Для анализа спектра молекулярной массы выделенной ДНК, для анализа результата действия рестриктаз на ДНК, а также для оценки проведения полимеразной цепной реакции используют метод электрофореза. Поскольку любая молекула ДНК в водном растворе отрицательно заряжена, появляется возможность разделить смесь фрагментов ДНК различных размеров по их длине с помощью электрофореза. ДНК помещают в гель (обычно, агарозный для относительно длинных и сильно отличающихся молекул или полиакриламидный для электрофореза с высоким разрешением), который помещают в постоянное электрическое поле. Из-за этого молекулы ДНК будут двигаться к положительному электроду (аноду), причем их скорости будут зависеть от длины молекулы: чем она длиннее, тем сильнее ей мешает двигаться гель и, соответственно, тем ниже скорость. После электрофореза смеси фрагментов разных длин в геле образуют полосы, соответствующие фрагментам одной и той же длины. С помощью маркеров (смесей фрагментов ДНК известных длин – лунка №1 на рис.) можно установить длину молекул в образце.
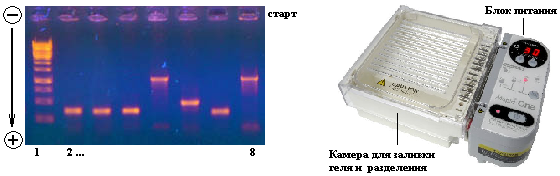
Визуализовать результаты фореза можно двумя способами. Первый, наиболее часто используемый в последнее время — добавление в гель веществ, флуоресцирующих в присутствии ДНК (традиционно использовался довольно токсичный бромистый этидий; в последнее время в обиход входят более безопасные вещества). Бромистый этидий светится оранжевым светом при облучении ультрафиолетом, причем при связывании с ДНК интенсивность свечения возрастает на несколько порядков (рис. 4). Другой метод заключается в использовании радиоактивных изотопов, которые необходимо предварительно включить в состав анализируемой ДНК. В этом случае на гель сверху кладут фотопластинку, которая засвечивается над полосами ДНК за счет радиоактивного излучения (этот метод визуализации называют авторадиографией).
Кроме «обычного» электрофореза в пластине из геля, в некоторых случаях используют капиллярный электрофорез, который проводят в очень тонкой трубочке, наполненной гелем (обычно полиакриламидным). Разрешающая способность такого электрофореза значительно выше: с его помощью можно разделять молекулы ДНК, отличающиеся по длине всего на один нуклеотид.
Задача.
Провести разделение по молекулярной массе выделенной в работе №3 ДНК из культуры фибробластов методом электрофореза в агарозном геле.
Протокол работы.
Перед проведением собственно электрофореза, необходимо приготовить следующие растворы:
Концентрированный буфер для электрофореза (50x, pH 8,5): 242 г Трис, 57,1 мл ледяной уксусной кислоты, 100 мл 0.5M EDTA. Довести водой до 1 литра.
Раствор красителя Cresol red: 50mM в H2O.
Буфер для нанесения: 0,25 мл 0,5% лаурилсульфата натрия (SDS), 1 мл 0,1М EDTA (pH 8,0), 2,5 мл 50% глицерола, 1,25 мл воды (общий объем 5 мл). Добавить раствор красителя Cresol red до конечной его концентрации 0.1mM.
Приготовить гель для электрофореза. Готовят 1% агарозный гель в 1 х буфере для электрофореза и охлаждают его до 70 0С. Для инактивации РНКаз добавляют в гель сухой йодацетат натрия до конечной концентрации 10 мМ. Охлаждают смесь до примерно 50 0С, выливают в кювету для электрофореза (слой толщиной 3 мм) и оставляют на 30 мин при комнатной температуре. После застывания геля, заливают камеру буфером для электрофореза.Во избежание повреждения геля не лить буфер на гель и вытаскивать гребенку только когда буфер будет налит.
Этапы работы:
Образец (примерно 20 мкг) двухцепочечной ДНК растворяют в 3 мкл воды, тестированной на ДНК-азы (или берут 3-5 мкл жидкого ДНК-содержащего образца).
Денатурируют ДНК прогреванием при 100 °С в течение 1 минуты.
Добавляют к каждому образцу ДНК 2,5 мкл буфера для нанесения, осторожно перемешивают.
Вносят образцы в лунки агарозного геля и проводят электрофорез при градиенте напряженности 2 В/см в 1 х буфере для электрофореза. Во время электрофореза должна быть обеспечена рециркуляция буфера, чтобы его рН поддерживался на уровне рН 8,5.
Когда Cresol red дойдет до конца геля, электрофорез останавливают и окрашивают гель бромистым этидием (0,5 мкг/мл в 25 мМ трис-HCl, рН 9,0) в течение 1 ч при осторожном покачивании. При сильном фоновом окрашивании лишний бромистый этидий можно отмыть водой (две смены по 30 мин каждая).
Просматривают гель в УФ-трансиллюминаторе и фотографируют его.