

3. 5. 4 Титрование солей
Метод нейтрализации применяется не только для титрования кислот и оснований но и для титрования солей, обладающих свойствами слабых кислот или оснований. Так, например, раствор соды может быть оттитрован раствором сильной кислоты, поскольку сода имеет свойства слабого основания. При титровании соды – карбоната натрия происходят следующие превращения:
![]() при
добавлении половины кислоты, требующейся
для титрования.
при
добавлении половины кислоты, требующейся
для титрования.
При
дальнейшем титровании идет реакция:
![]()
 V,
см3
V,
см3
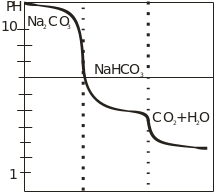
Рисунок 5- Кривая титрования соды хлористоводородной кислотой
В связи с этим карбонат натрия можно оттитровать хлористоводородной кислотой двумя способами. 1. Хлористоводородной кислотой титруют карбонат до бикарбоната в присутствии индикатора, имеющего интервал перехода (8-10), фиксируют VHCL. Далее титруют бикарбонат до pH 3.75 в присутствии другого индикатора (pH 3.1-4.4). Через объем находят содержание соды.
2. Карбонат натрия можно оттитровать сразу до II т.э. с метиловым оранжевым. Образуется угольная кислота, которая сразу разлагается и не мешает титрованию. Точку эквивалентности устанавливают различными способами. Одним из наиболее широко применяемых способов фиксации т.э. является индикаторный метод. При этом методе конечную точку титрования определяют по изменению цвета раствора в присутствии индикатора.
Лекция 4 Кислотно-основные индикаторы. Титрование в неводных средах. Теория кислоти оснований.
4. 1 Кислотно-основные индикаторы
В 1894г Оствальдом была создана, так называемая ионная теория индикаторов. Согласно этой теории кислотно-основные индикаторы – это сложные органические вещества (слабые органические кислоты или основания: HInd или IndOH), способные изменять свою окраску в зависимости от pH раствора. Известно около 200 кислотно-основных индикаторов, относящихся к различным классам органических соединений. Кроме индивидуальных для титрования применяют смешанные индикаторы, представляющие собой смеси 2, 3 и более индикаторов, которые дают более четкие переходы окраски при изменении pH раствора.
В растворах индикаторы могут существовать в молекулярной и ионной формах. Эти формы окрашены в разный цвет и находятся в равновесии, которое зависит от pH среды.
![]()
Например, кислотный индикатор метилоранж, в молекулярной форме имеет красную окраску, а в нейтральной и щелочной среде – жёлтую. Изменение кислотности раствора приводит к смещению равновесия диссоциации либо вправо либо влево, что сопровождается изменением окраски раствора.
Предложенная
позже хромофорная
теория связывает
изменение окраски индикаторов с
изменением строения индикаторов в
результате внутримолекулярной
перегруппировки. Свое название эта
теория получила из-за того, что окраска
органических соединений приписывается
наличию в них особых групп, называющихся
хромофорами. К хромофорам относятся
группы:
![]() ,
азогруппа –N=N-,
переходящая в группу =N-NH-,
группа =С=0. Вызванная хромофорами окраска
соединения усиливается присутствием
в молекуле соединения групп, называемых
ауксохромами. Важнейшими ауксохромами
являются группы –OH и –NH2,
а также их производные, например –N(CH3)2,
-N(C2H5)2
и т.д. Ауксохромы сами по себе не способны
придавать окраску соединению, но
присутствуя с хромофорами, они усиливают
действие последних. Если в результате
внутримолекулярной перегруппировки в
индикаторе возникают или исчезают
хромофорные или ауксохромные группы,
влияющие на окраску, то окраска
изменяется.Ионная и хромофорная теории
не исключают, а дополняют друг друга.
Ионизация молекул индикатора обычно
приводит к внутримолекулярной
перегруппировке и изменению окраски.
При изменении pH
раствора все ислотно-основные индикаторы
изменяют свою окраску не скачкообразно,
а плавно, т.е. в определенном интервале
значений pH.
Этот интервал называется интервалом
перехода индикатора
,
азогруппа –N=N-,
переходящая в группу =N-NH-,
группа =С=0. Вызванная хромофорами окраска
соединения усиливается присутствием
в молекуле соединения групп, называемых
ауксохромами. Важнейшими ауксохромами
являются группы –OH и –NH2,
а также их производные, например –N(CH3)2,
-N(C2H5)2
и т.д. Ауксохромы сами по себе не способны
придавать окраску соединению, но
присутствуя с хромофорами, они усиливают
действие последних. Если в результате
внутримолекулярной перегруппировки в
индикаторе возникают или исчезают
хромофорные или ауксохромные группы,
влияющие на окраску, то окраска
изменяется.Ионная и хромофорная теории
не исключают, а дополняют друг друга.
Ионизация молекул индикатора обычно
приводит к внутримолекулярной
перегруппировке и изменению окраски.
При изменении pH
раствора все ислотно-основные индикаторы
изменяют свою окраску не скачкообразно,
а плавно, т.е. в определенном интервале
значений pH.
Этот интервал называется интервалом
перехода индикатора
![]() .
Каждый индикатор имеет свой интервал
перехода, который зависит от особенностей
структуры индикатора. Интервал перехода
окраски индикатора характеризуется
показателем титрования pT.
Показатель титрования – это значение
pH,
при котором наблюдается наиболее резкое
изменение цвета индикатора.
.
Каждый индикатор имеет свой интервал
перехода, который зависит от особенностей
структуры индикатора. Интервал перехода
окраски индикатора характеризуется
показателем титрования pT.
Показатель титрования – это значение
pH,
при котором наблюдается наиболее резкое
изменение цвета индикатора.
Интервал значений pH, в котором происходит изменение окраски индикатора обозначают :
![]() ,
,
где Кинд – константа диссоциации индикатора
Значение К, окраска и приведены в химических справочниках.
Таблица 1- Окраска индикаторов
Индикатор |
Окраска |
pT |
|
|
В кислотной форме |
В основной форме |
|||
фенолфталеин |
бесцветная |
розовая |
9,0 |
8,2-10 |
метилоранж |
красная |
желтая |
3,2 |
3,1-4,4 |
Индикаторы применяют или в виде растворов, или в виде индикаторных бумаг.
4. 2 Теория кислот и оснований
Содержание понятий «кислот» и «основание» в процессе развития химической науки существенно менялось, оставаясь одним из основных вопросов химии. Одной из первых теорий кислот и оснований является теория Аррениуса. Согласно определению Аррениуса-Оствальда кислоты – это вещества, диссоциирующие в воде с образованием иона водорода H+, а основания – вещества, дающие анион гидроксила OH-. По мере накопления данных, развития теории растворов оказалось, что многие вещества, не имеющими в своем составе H+ или OH- обладают свойствами кислот или оснований. Было доказано, что в свободном виде H+ вообще не существует. В водных растворах эти ионы гидратированы, а в неводных сольватированы. Так, например:
![]()
Исследования показали, что некоторые соли в неводных растворителях ведут себя как кислоты или основания. Так например KNH2 в растворе аммиака ведет себя как KOH в воде, т.е. является сильным основанием. Он окрашивает фенолфталеин, обладает электропроводностью, нейтрализует кислоты. Другая соль NH4Cl ведет себя в сухом аммиаке как HCl, т.е. является сильной кислотой. Следовательно, основные и кислотные свойства присущи не только соединениям, имеющим ионы водорода и гидроксильные группы. Поэтому следующей теорией кислот и оснований стала теория сольвосистем.
Согласно этой теории кислотами и основаниями являются химические соединения, образующие катионы и анионы, идентичные катионам и анионам данного растворителя.
Так, например жидкий аммиак диссоциирует:
![]()
![]() ,
,
значит NH4Cl – кислота (такой же катион)
![]() -
основание (такой же анион).
-
основание (такой же анион).
Недостатком этой теории является то, что в некоторые растворители не диссоциируют ни на катионы ни на анионы, а кислоты и основания в них существуют.
Протолитическая теория Бренстеда-Лоури.
Согласно этой теории кислотами являются химические соединения, способные отдавать протоны другим веществам, а основаниями – вещества, способные присоединять протоны.
Кислотами могут быть и молекулы и катионы и анионы. Например, вода:

![]()
Таким
образом, каждая кислота имеет сопряженное
основание (![]() ),
а каждое основание имеет сопряженную
кислоту.
),
а каждое основание имеет сопряженную
кислоту.
Сила кислот и оснований зависит от природы растворителя. Так, например, в растворе жидкого аммиака все кислоты полностью диссоциированны т.к. жидкий аммиак проявляет свойства основания. В воде, менее сильном основании, не все кислоты диссоциируют, а лишь только сильные неорганические.
К недостаткам теории Бренстеда-Лоури относится то, что эта теория исключает возможность проявления кислотного характера веществами, не содержащими водород. Поэтому наряду с этой теорией появилась еще одна теория – электронная теория Льюса.
Согласно этой теории основанием является вещество, обладающее неподеленной свободной парой электронов. Например, аммиак является основанием, т.к. его молекула имеет неподеленную электронную пару.
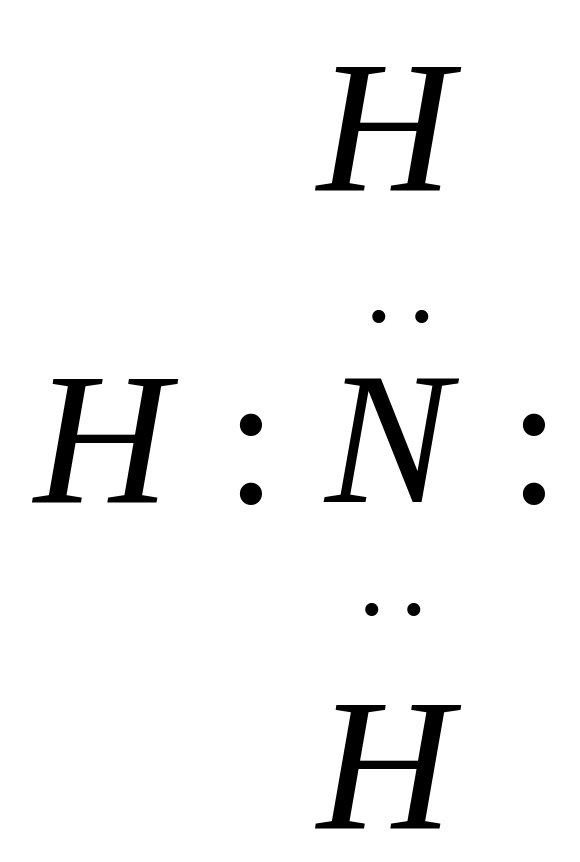
Кислотой является вещество, в молекуле которого не хватает пары электронов до образования устойчивой электронной группировки. Например: BCl3
![]()
По теории Льюиса вещество не обязательно должно иметь H+ чтобы обладать кислотными свойствами. Так, NH3 и BCl3 взаимодействуют с образованием соли:
+
или NH3+HClNH4Cl
Электронная теория значительно расширила понятие о кислотах и основаниях. Недостатком этой теории является то, что в ней не объясняется тот факт, что одно и то же вещество может быть и кислотой и основанием в зависимости от природы растворителя. В настоящее время на основании исследований ряда ученых было доказано, что одно и то же вещество в зависимости от растворителя, в котором оно растворено, может быть отнесено к кислотам или основаниям.
Современная теория кислот и оснований.
Эта теория дает следующее определение кислотам и основаниям:
«Кислота – это вещество, которое является донором протонов или акцептором электронной пары или дающее такой же катион лиония, как и растворитель, в котором оно растворено. Основание – это вещество, являющееся акцептором протонов, или донором электронной пары, или дающее такой же анион лиата, как и растворитель, в котором оно растворено.
Например соль CH3COONa диссоциирует в уксусной кислоте согласно уравнению:
CH3COONa CH3COO-+Na+ (основные свойства)
Следовательно, CH3COONa можно количественно оттитровать какой- либо сильной кислотой, например, хлорной:
HClO4+CH3COONaNaClO4+CH3COOH.
4. 3 Титрование в неводных средах.
Химическая теория растворов Д. И. Менделеева рассматривает растворитель не тольао как среду, в которой протекает реакция, но и как непосредственного участника химиического процесса. Согласно теории неводных сред, разработанной нашими учеными Измайловым и Крешковым одно и то же вещество может вести себя по разному в зависимости от растворителя, т.е. сила кислот и оснований зависит от природы растворителя.
При классификации по донорно-акцепротным свойствам обычно выделяют протонные и апротонные растворители. Притонные могут отдавать или принимать протон и таким образом участвовать в процессе кислотно-основного взаимодействия. Апротонные растворители не проявляют кислотно-основных свойств и не вступают в протолитическое равновесие с растворённым веществом. Протонные растворители принято подразделять на:
1. Амфотерные растворители.Это такие растворители, которые играют роль основания по отношению к кислотам и роль кислот по отношению к основаниям. Эти растворители отличаются способностью и отдавать и присоединять протоны. К ним относятся: H2O, CH3OH, C2H3OH и другие.
Кислые растворители. Это вещества кислого характера, молекулы которого могутлишь отдавать протоны. HF, H2SO4, CH3COOH и другие.
3. Основные растворители. Это вещества, обладающие ярко выраженным сродством к протонам (NH3, N2H4).
По влиянию на кислотно-основные свойства растворённого вещества растворители принято делить на нивелирующие и дифференцирующие.
Нивелирующие – это растворители, в которых кислоты и основания раздельной природы не меняют соотношения в своей силе (вода, уксусная кислота и др.)
Дифференцирующие – растворители, в которых кислоты и основания заметно изменяют соотношение в своей силе (ДМФ, ацетон и др).
К нивелирующим растворителям относятся или очень сильные кислоты или очень сильные основания, например CH3COOH – гидразин. Поскольку это сильные кислоты или основания, все кислоты в их среде становятся одинаковыми по своей силе, то же касается и оснований.
К дифференцирующим же относятся растворы, в среде которых проявляются значительные различия в силе кислот и оснований. Например, ДМФ, ДМСО, пиридин, ацетон. В среде этих растворителей можно раздельно оттитровать не только 2-х, 3-х, но и даже 5 и 6-и компонентные смеси.
Используя влияние неводных растворителей на свойства растворенных электролитов, можно проводить кислотно-основное титрование в неводных средах таких веществ, которые не могут быть оттитрованы в воде. Так, например, многие соли в воде проявляют свойства очень слабых или кислот или оснований и не могут быть оттитрованы непосредственно основаниями или кислотами. В неводных же средах их кислотность или основность повышается настолько, что их можно количественно оттитровать кислотой или основанием.
Титрование в неводных средах получило широкое применение в аналитической химии. Это связано со следующими причинами.
В неводных средах можно оттитровать те вещества, которые в воде не растворяются.
В неводных средах можно титровать те вещества, которые в воде не дают резких конечных точек титрований.
В неводных средах можно проводить не только к/о, но и о/в, комплекснометрическое, осадительное титрование.
Лекция 5 Окислительно-восстановительные методы (редоксиметрия).
1 Суть редоксиметрического метода анализа
Этот метод основан на использовании окислительно-восстановительных реакций. В качестве титрантов применяют растворы окислителей или восстановителей. Как правило, окислителями титруют вещества, которые могут окисляться, а восстановителями вещества, которые могут восстанавливаться. С помощью этого метода можно определять и неорганические и органические вещества, способные к окислению или восстановлению.
Существуют несколько способов титрования: прямой и обратный.
В процессе титрования изменяется не рН раствора, а его окислительно-восстановительный потенциал. Если реакцию между окислителем и восстановителем представить в виде:
![]() ,
,
то константу равновесия можно представить следующим образом:
![]() .
.
Воспользовавшись уравнением Нернста, можно выразить концентрации окислителя и восстановителя через потенциалы. После преобразований получим выражение для константы равновесия:
![]() .
.
Таким
образом, чем больше разность между
стандартными потенциалами окислителя
и восстановителя, тем больше константа
равновесия. Следовательно, тем более
вероятно, что реакция идет до конца.Поэтому
для титрования выбирают сильные
окислители и сильные восстановители,
имеющие высокие значения стандартных
потенциалов. К сольным окислителям
относятся
![]() .
К сильным восстановителям относятся
растворы ионов металлов,
.
К сильным восстановителям относятся
растворы ионов металлов,
![]() .
.
5. 2 Кривые титрования в редоксиметрии
В
процессе титрования меняется Е раствора,
поэтому такую зависимость можно выразить
графически. Например, рассмотрим, как
изменяется потенциал раствора
![]() при титровании этих ионов титрантом
при титровании этих ионов титрантом
![]() .
Запишем реакцию:
.
Запишем реакцию:
![]() .
.
Согласно уравнению Нернста до точки эквивалентности потенциал раствора рассчитывают по формуле:
 ,
,
после точки эквивалентности:
 .
.
На рисунке 1 изображена кривая титрования кривая титрования раствора FeSO4 раствором КМп04.
Кривые окислительно-восстановительного титрования выглядят, в общем, как и кривые титрования кислот и оснований. В близи точки эквивалентности они имеют резкий скачок потенциала. Поэтому для фиксирования точки эквивалентности можно воспользоваться индикаторами, которые меняют свой цвет в зависимости от потенциала системы. В отличие от кривой кислотно-основного титрования скачок не зависит от разбавления и его можно повысить, если один из образующихся ионов связывать в комплекс.
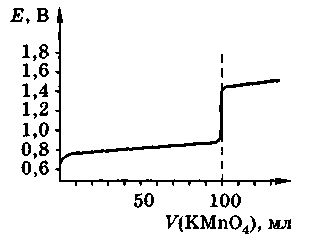
Рисунок 1- Кривая титрования 100,0 см3 0,lMFeSO4 0,1н. раствором КМп04.
5. 3 Индикаторы, применяемые в редоксиметрии
В окислительно-восстановительном титровании точку эквивалентности можно определить тремя способами:
1.
При титровании часто можно обойтись
вообще без индикаторов. Безиндикаторное
титрование возможно в том случае, если
титрант или определяемый раствор имеют
яркую окраску, как, например, в случае
титрования
![]() перманганата калия. Как известно, раствор
перманганата калия. Как известно, раствор
![]() яркого малиново-фиолетового цвета. В
результате восстановления
яркого малиново-фиолетового цвета. В
результате восстановления
![]() образуются бесцветные ионы
образуются бесцветные ионы![]() .
Без индикатора можно также титровать
раствором йода, поскольку
.
Без индикатора можно также титровать
раствором йода, поскольку
![]() имеет темную окраску, а
имеет темную окраску, а
![]() бесцветен.
бесцветен.
С помощью индикаторов.
Индикаторы в редоксиметрии можно разделить на две группы:
1)
Индикаторы, которые вступают в
специфическую реакцию с избытком
окислителя или восстановителя. Например
ионы
![]() дают ярко-розовый комплекс с
дают ярко-розовый комплекс с
![]() поэтому, если в растворе появится хотя
бы одна капля
,
весь раствор окрашивается в розовый.
поэтому, если в растворе появится хотя
бы одна капля
,
весь раствор окрашивается в розовый.
Индикаторы, у которых перемена окраски не зависит от специфических свойств окислителя или восстановителя, а связана с достижением титруемым раствором определенного потенциала. Такие индикаторы называются окислительно-восстановительными. Окисленная и восстановленная формы имеют различную окраску.
Их превращение можно представить следующим образом:
![]() ,
,
где
![]() – окисленная форма;
– окисленная форма;
![]() – восстановленная.
– восстановленная.
Применяя к таким индикаторам уравнение Нернста, получим:
![]() .
.
Таким образом, при изменении потенциала раствора изменяется соотношение между окисленной и в восстановленной формами. Если к окислительно-восстановительной системе прилить 1-2 капли индикатора, то установится соответствующие потенциалу системы соотношение между концентрациями окисленной и восстановленной форм индикатора. При этом раствор приобретает соответственную окраску. Для любой системы можно подобрать такой индикатор, у которого изменение окраски индикатора происходит вблизи точки эквивалентности.
5. 4 Примеры окислительно-восстановительных методов титрования.
5. 4. 1 Перманганатометрия
Перманганатометрией называют метод, в котором рабочим раствором, т.е. титрантом, является раствор перманганата калия . Определяемыми веществами являются катионы металлов, способные к окислению.
В зависимости от условий, в которых протекает реакция окисления-восстановления анион может принимать различное количество электронов:
Среда: |
Кислая |
|
|
Нейтральная |
|
|
|
Щелочная |
|
|
В кислой среде окислительно-восстановительный потенциал системы самый большой, поэтому окисление перманганатом калия с аналитическими целями проводят в кислой среде. В связи с этим основное уравнение перманганатометрии имеет вид:
![]() ;
;
![]() ;
;
![]() ;
;
Обычно
готовят 0,1н. раствор
или 0,05н.
.
Перманганат калия, применяемый для
приготовления рабочего раствора, как
правило, содержит ряд примесей, из
которых наиболее значимые примеси
![]() .
Кроме того, концентрация перманганата
постоянно меняется, т.к. все время идет
его восстановление примесями органических
веществ, которые находятся в воздухе и
дистиллированной воде. Поэтому
концентрацию
устанавливают по стандартному веществу,
концентрация которого точно известна
и не меняется. Первичным стандартом в
перманганатометрии являются такие
вещества как оксалат аммония, натрия
или щавелевая кислота:
.
Кроме того, концентрация перманганата
постоянно меняется, т.к. все время идет
его восстановление примесями органических
веществ, которые находятся в воздухе и
дистиллированной воде. Поэтому
концентрацию
устанавливают по стандартному веществу,
концентрация которого точно известна
и не меняется. Первичным стандартом в
перманганатометрии являются такие
вещества как оксалат аммония, натрия
или щавелевая кислота:
![]() .
.
Взаимодействие щавелевой кислоты с перманганатом калия протекает согласно уравнению:
![]() .
.
Разность окислительно-восстановительных потенциалов:
![]() .
.
Большая разность потенциалов показывает, что реакция идет до конца. Однако скорость прямой реакции мала и реакция идет очень медленно. На скорость прямой реакции влияют следующие факторы: рН, температура, катализатор. Поэтому для ускорения реакции повышают рH раствора (в кислой среде E0 имеет максимально значение). Реакцию проводят при нагревании (70-800С). Катализатором этой реакции являются ионы двухвалентного марганца. Они появляются в результате реакции окисления и по мере накопления течение реакции ускоряется до точки мгновенного взаимодействия.
Титрование перманганатом проводят без индикатора, т.к. раствор сам имеет малиновую окраску и в точке эквивалентности лишняя капля титранта окрашивает раствор в розовый цвет.
Перманганатометрия используется для определения содержания как восстановителей, так и окислителей. Из окислителей этим методом наиболее часто определяют ионы двухвалентного железа. Соединения двухвалентного железа легко определяются в кислой среде:
![]() .
.
При
окислении ионы двухвалентного железа
переходят в ионы трехвалентного железа,
поэтому
![]() ,
,
![]() .
Реакция идет быстро даже без нагревания,
а лучше ее проводить при охлаждении и
среде инертного газа для предотвращения
окисления ионов железа кислородом
воздуха.
.
Реакция идет быстро даже без нагревания,
а лучше ее проводить при охлаждении и
среде инертного газа для предотвращения
окисления ионов железа кислородом
воздуха.
При анализе сплавов железа, железной руды и минералов, где железо находится как в двухвалентном, так и в трехвалентном виде, предварительно восстанавливают трехвалентное железо в двухвалентное, а затем уже оттитровывают перманганатом. Восстановления трехвалентного железа проводят разными способами: цинком, алюминием и др.
5. 4. 2 Йодометрия
Кроме перманганата, в оксидиметрии в качестве окислителя широко применяют йод:
![]() .
.
В
этой реакции каждый атом йода присоединяет
один электрон, и, следовательно, эквивалент
йода равен его атомной массе. Стандартный
окислительно-восстановительный
потенциал системы
![]() ,
т.е. немного меньше, чем у системы
,
т.е. немного меньше, чем у системы
![]() .
.
Вследствие этого йод окисляет гораздо меньшее число восстановителей по сравнению с перманганатом. Реакция окисления йода обратима, и ее направление определяется условиями, в которых она протекает. Наибольший окислительно-восстановительный потенциал этой системы проявляется в нейтральной среде. В щелочных и кислых средах эта реакция протекает по другому механизму. Особенностью йодометрии является тот факт, что в качестве рабочего раствора, т.е. титранта раствор йода используют крайне редко. Раствором нельзя непосредственно титровать какой-то восстановитель, как это делают в пермангаматометрии. Это связано с тем, что – летучее вещество, которое быстро улетучивается из бюретки, кроме того, он разлагается на свету. Поэтому в йодометрии используют метод обратного титрования. Суть метода заключается в том, что титрантом является не сам , а раствор первичного стандарта, например тиосульфат Na.
Эта реакция протекает согласно уравнению:
![]() ,
,
при этом ионы окисляются:
![]() и
и
![]() .
.
При титровании в бюретку помещают раствор тиосульфата натрия, а в конические колбы для титрования – определенный объем раствора , приготовленный из точной навески.
Концентрацию
тиосульфата можно установить и по другим
окислителям, например, по
![]() .
В качестве индикатора в этом титровании
используют водный раствор крахмала.
Его использование основано на том, что
раствор крахмала окрашивается йодом в
темно-синий цвет. В точке эквивалентности
синяя окраска раствора исчезает, и
раствор становится бесцветным.
Йодометрическое титрование используется
для определения содержания как
окислителей, так и восстановителей,
можно использовать как прямую йодометрию
так и обратную.
.
В качестве индикатора в этом титровании
используют водный раствор крахмала.
Его использование основано на том, что
раствор крахмала окрашивается йодом в
темно-синий цвет. В точке эквивалентности
синяя окраска раствора исчезает, и
раствор становится бесцветным.
Йодометрическое титрование используется
для определения содержания как
окислителей, так и восстановителей,
можно использовать как прямую йодометрию
так и обратную.
5. 4. 3 Хроматометрия
В
качестве окислителей в
окислительно-восстановительных методах
широко используют раствор дихромата
калия. Метод основанный на применении
этого окислителя, называется хроматометрией.
Дихромат калия отличается от других
окислителей очень высокой устойчивостью,
поэтому его титр и нормальность не
изменяются в течении нескольких месяцев.
Готовят раствор дихромата калия по
точной навеске химически чистого
препарата в мерной колбе, т.е. первичный
стандарт в данном случае не требуется.
Точку эквивалентности в хроматометрии
определяют при помощи индикатора
дифениламина, который в точке
эквивалентности изменяет свой цвет.
Дифениламин является характерным
представителем окислительно-восстановительных
индикаторов. Хроматометрию наиболее
часто применяют для определения ионов
![]() и для определения общего содержания
железа в его сплавах, рудах и минералах.
Хроматометрия используется для
определения других катионов металлов,
способных к восстановлению. Кроме того,
используя метод обратного титрования,
можно с помощью этого метода определять
и содержание окислителей в образцах.
и для определения общего содержания
железа в его сплавах, рудах и минералах.
Хроматометрия используется для
определения других катионов металлов,
способных к восстановлению. Кроме того,
используя метод обратного титрования,
можно с помощью этого метода определять
и содержание окислителей в образцах.
5. 4. 4 Броматометрия и бромометрия.
В
качестве окислителей в редоксиметрии
часто применяют или бромат калия
![]() или смесь бромата и бромида (
или смесь бромата и бромида (![]() ).
Окисление ведут в кислой среде, при этом
определяемые ионы окисляются до высшей
степени окисления, а бромат и бромид
восстанавливаются до
).
Окисление ведут в кислой среде, при этом
определяемые ионы окисляются до высшей
степени окисления, а бромат и бромид
восстанавливаются до
![]() .
Выделившийся бром обнаруживают или по
появлению желтой окраски раствора или
по изменению цвета индикаторов. С помощью
бромо- и броматометрии определяют
содержание ионов мышьяка, сурьмы, а
также фенола, анилина, различных
производных бензола, способных к
окислению.
.
Выделившийся бром обнаруживают или по
появлению желтой окраски раствора или
по изменению цвета индикаторов. С помощью
бромо- и броматометрии определяют
содержание ионов мышьяка, сурьмы, а
также фенола, анилина, различных
производных бензола, способных к
окислению.
5. 5. 5 Цериметри
В
качестве окислителя могут быть
использованы соли
![]() .
Это связано с тем, что ионы четырехвалентного
церия легко восстанавливаются до
.
Это связано с тем, что ионы четырехвалентного
церия легко восстанавливаются до
![]() .
В результате происходит обесцвечивание
желтого раствора соли
.
В результате происходит обесцвечивание
желтого раствора соли
![]() ,
т.к. соли
желтые,
бесцветные. Такое титрование, как и в
случае с перманганатом калия, можно
проводить без индикатора. Цериметрию
можно использовать для тех же случаев,
что и перманганатометрию, только эти
соли церия отличаются большей
устойчивостью.
,
т.к. соли
желтые,
бесцветные. Такое титрование, как и в
случае с перманганатом калия, можно
проводить без индикатора. Цериметрию
можно использовать для тех же случаев,
что и перманганатометрию, только эти
соли церия отличаются большей
устойчивостью.
Лекция 6 Метод комплексообразования (комплексометрия)
6. 1 Общаяя характеристика метода
Комплексометрия основана на реакциях образования комплексов. В самом общем смысле под комплексом (комплексным соединением) в химии понимают сложную частицу, состоящую из составных частей, способных к автономному существованию. Можно отметить основные признаки, позволяющие выделить комплексные соединения в особый класс химических соединений:
- способность отдельных составных частей к самостоятельному существованию;
- сложность состава;
- частичная диссоциация на составные части в растворе по гетеролитическому механизму;
- наличие положительно заряженной центральной частицы – комплексообразователя (обычно это ион металла), связанной с лигандами;
- наличие определенной устойчивой пространственной геометрии расположения лигандов вокруг комплексообразователя. Примеры:
Комплекс Составные части
Ni(NH3)62+ Ni2+ , NH3
[Co(NH3)6]SO4 [Co(NH3)6]2+ , SO42- , Co2+ , NH3
Лиганды («зубчатые структуры») могут быть бидентатными, монодентатными, полидентатнами. Дентатностью называется число донорных атомов лиганда, образующих координационные связи с центральным атомом.
Известно много монодентатных неорганических и органических лигандов, однако их применению в комплексометрии препятствует то, что ступенчатые константы устойчивости соответствующих комплексов мало различаются между собой. Поэтому при увеличении количества добавленного лиганда концентрация ионов металла изменяется постепенно и кривая титрования не имеет скачка.
Хелаты (хело-«клешня») – соединения, которые охватывают центральный ион. Важнейшая особенность хелатов – их повышенная устойчивость по сравнению с аналогично построенными нециклическими комплексами. Именно поэтому полидентатные лиганды и хелатные комплексы нашли широкое применение в аналитической химии.
Скорость комплексообразования имеет большое значение в аналитической химии. Например, при прямом комплексонометрическом титровании реакция определяемого иона с титрантом должна протекать практически мгновенно, иначе индикация конечной точки титрования существенно затрудняется.
Устойчивость комплекса определяется как фундаментальными факторами (природой комплексообразователя и лигандов), так и внешними условиями (температурой, природой растворителя, ионной силой, составом раствора).
Таким образом, комплексометрия основана на применении реакции образования комплексов Ме с аминополикарбоновыми кислотами. В результате реакции образуются внутрикомплексные соли (хелатные соли).
Комплексонами
обычно называют органические соединения,
представляющие собой производные
аминополикарбоновых кислот. Простейший
комплексон – нитрилотриуксусная
кислота, известная под названием
комплексон
1![]() .
.
Наибольшее значение имеет четырехосновная этилендиаминтетрауксусная кислота (ЭДТА) –комплексон 2.
Комплексоны наряду с карбоксильными группами содержат аминный азот. Благодаря такому строению эти соединения отличаются полидентантностью, т.е. способностью образовывать сразу несколько координационных связей с ионами металлов. На практике обычно применяют двунатриевую соль этилендиаминтетрауксусной кислоты, которую называют комплексоном 3.или трилоном Б. комплексон 3.
Комплексон 3 образует со многими катионами устойчивые малодиссоциированные растворимые в воде внутрикомплексные соли. Комплексоны широко применяются для качественного определения многих катионов.
Комплексон 1 – нитрилотриуксусная кислота (НТА, условно (X3Y):
C H2COOH
H2COOH
H OOCCH2
– N
OOCCH2
– N
CH2COOH или
|
(дентат. = 4); |

Комплексон 2 - этилендиаминтетрауксусная кислота (ЭДТА, условно XH4Y):
|
(дентат. =6); |

Комплексон 3- дигидрат двунатриевой соли этилендиаминтетрауксусной кислоты (условно Na2H2Y).
Комплексоны 1,2 и 3 сильные органические кислоты, диссоциируют ступенчато, причем степень диссоциации зависит от pH.
Так, комплексон 3 в воде диссоциирует согласно уравнениям:
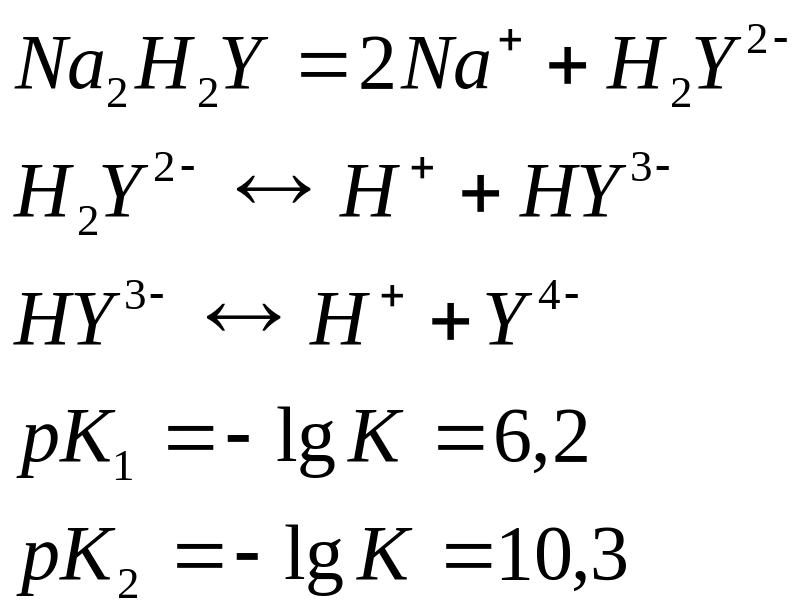
Комплексон 2 диссоциирует:

В
сильно кислых растворах с pH<3
образуются менее устойчивые комплексные
соединения. В сильнощелочных растворах
с pH>10
могут выпадать осадки гидроокисей
металоов. Поэтому при комплексометрическом
титровании нужно поддерживать pH
в интервале от 3 до 10. При этом для каждого
катиона существует свое строго
определенное значение pH,
при котором комплекс наиболее устойчив.
Поэтому для поддержания определенного
значения pH
готовят буферные
растворы.
Титрование большинства катионов обычно
проводят в аммиачной буферной среде
![]() при pH=8-9.
В кислой же среде только некоторые
катионы образуют прочные комплексы с
комплексонами.
при pH=8-9.
В кислой же среде только некоторые
катионы образуют прочные комплексы с
комплексонами.
6. 2 Комплексонометрическое титрование.
Суть комплексометрического титрования заключается в следующем. Титрантом является комплексон 2, реже 3. В раствор, в котором содержится определяемый ион добавляют определенное количество металлиндикатора. Металлиндикатор образует с определяемым ионом Ме окрашенный комплекс. Раствор начинают титровать комплексоном 3. Комплекс с металлоиндикатором постепенно разделяется и образуется новый, более устойчивый комплекс Ме с комплексоном – другого цвета. В точке эквивалентности первоначальный цвет соединения Ме с индикатором исчезает и появляется окраска, свойственная свободному индикатору.
Таким образом, комплексонометрия (хелатометрия), представляет собой титриметрический метод анализа, основанный на использовании реакций ионов металлов с комплексонами, сопровождающихся образованием устойчивых малодиссоциирующих внутрикоплексных солей.
При титровании комплексоном 3 солей протекают следдующие реакции: реакция1
Комплексоны вступают в реакции с катионами многих металлов в отношении 1:1, образуя малодиссоциирующие растворимые в воде комплексы, соли, называемые комплексонатами:

Катионы Mg2+, Ca2+, Sr2+, Ba2+, Al3+, Mn2+, Cd2+, Zn2+, Fe2+, Co2+, Ni2+, Cu2+ образуют с комплексонами в кислой и нейтральной средах соединения состава H[Me2+X3-] или [Me2+X3-].
Например, катионы кальция связываются с комплексоном 3 по уравнению:
Ca2+ + Na2H2Y Na2[ CaY ] + 2H+.
Комплексные соли комплексона 3 (трилона Б) с металлами называют соответственно трилонатами. Внутрикомплексные ионы, образуемые трилоном Б с ионами Me2+, имеют следующее строение:

Трилон Б образует прочные растворимые в воде комплексные соединения с большим числом катионов металлов путем замещения атомов водорода карбоксильных групп на металл и координации последнего с азотом.
Прочность трилонатов очень велика.Так, например, константа устойчивости трилоната никеля равна NiY2-= 4,2. 1018. На устойчивость этих внутрикомплексных ионов мало влияют температура и органические растворители. При всех значениях pH катионы металлов любой степени окисления реагируют в отношении 1:1. На основании величин констант нестойкости комплексов сделаны выводы, что у двухзарядных катионов трилонаты устойчивы в щелочных и аммиачных растворах, а у трехзарядных - даже в кислых растворах. С увеличением степени окисления металла прочность комплексонатов (трилонатов) возрастает.
6. 3 Индикаторы, применяемые в комплексонометрии
В комплексонометрии чаще всего для фиксирования момента эквивалентности применяют индикаторы, меняющие свой цвет в зависимости от концентрации ионов металла в растворе, а поэтому называющиеся металлоиндикаторами. Они являются органическими красителями и в свободном состоянии окрашены. С катионами металлов они образуют внутрикомплексные интенсивно окрашенные соединения (отличающиеся по цвету от окраски индикатора в свободном состоянии).
HInd2- + Me2+ MeInd- + H+
синий красный
Если к раствору такого соединения добавить титрованный раствор комплексона III (трилона Б), то внутрикомплексное соединение металла с индикатором разрушается и образуется новое внутрикомплексное соединение, более прочное (трилонат). Освободившиеся молекулы (ионы) индикатора изменяют окраску раствора:
MeInd- + HI3- MeI2- + HInd- .
красный синий
Таким образом, в точке эквивалентности наблюдается переход от окраски раствора комплекса металла с индикатором (например, красной) к окраске самого индикатора. Если металл связан прочнее с индикатором, чем с комплексоном (т.е. устойчивость комплексоната меньше), то титрование невозможно (индикатор «блокирован»). Так как индикаторы изменяют окраску также и с изменением значений pH раствора, то титровать необходимо в буферных растворах.
Важнейшие индикаторы - эриохром черный Т и мурексид.
Эриохром черный Т (хромоген черный специальный ЕТ-00) - азокраситель, 0,0-диоксиазосоединение C20H13O7Na3S (условноH3Ind):
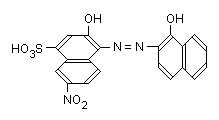
Индикатор диссоциирует с образованием Н+, Н2Ind-, HInd2-, Ind3-, поэтому эриохром чёрный Тспособен реагировать с ионами металла-комплексообразователя с образованием комплексных соединений. Сам индикатор окрашен в синий цвет. В нейтральной или щелочной среде при рН= 7...10 он образует с ионами металлов соединения красного цвета.
Чтобы окраска комплекса резко отличалась от окраски самого индикатора, титрование суказанным индикатором ведут при pH=9,3 (в присутствии аммиачного буфера) с образованием комплексных соединений красного цвета.
Мурексид (аммонийная соль пурпурной кислоты):
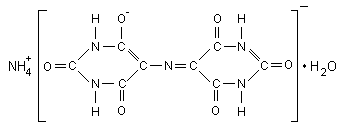
Мурексид представляет собой тёмно-красный порошок. Водный раствор мурексида окрашен в фиолетово-красный цвет, изменяющийся в зависимости от среды: при рН 9 – красно-фиолетовый, рН =9...10 –фиолетовый, рН 11 синефиолетовый. С катионами металлов мурексид образует комплексные растворимые в воде соединения красного или жёлтого цвета. Комплексы металлов с мурексидом имеют значительную устойчивость. Их прочность повышается с увеличением pH. Наиболее устойчивы комплексы с медью. С Ca2+ мурексид образует красный комплекс, с ионами Cu2+, Ni2+, Co2+ образует желтый комплекс.
При определении содержания ионов Ca2+ следует создавать значение pH > 12, так как при этом резче происходит переход окраски от красной к сине-фиолетовой. При определении содержания ионов Cu2+, Co2+ создают pH = 9 (аммонийным буфером), при этом происходит переход окраски от желтой к красно-фиолетовой.
Кроме того, применяют как индикаторы кислотный хром синий, кислотный темно-синий, сульфосалициловую кислоту и др.
6. 4 Способы комплексометрического титрования
В настоящее время комплексоны (и особенно трилон Б ) применяют в самых различных методах анализа - колориметрии, потенциометрии, гравиметрии и др. В титриметрическом анализе комплексоны применяют для определения общей жесткости воды, для анализа различных фармацевтических препаратов и т. д. Такое широкое распространение комплексонов, особенно, трилона Б связано прежде всего с тем, что из одной пробы анализируемого раствора можно определять до пяти катионов при различных значениях pH раствора, а также с большой точностью метода.
Приемы комплексометрического титрования могут быть различными: прямое титрование, обратное, титрование по методу вытеснения и алкалиметрическое.
6.4.1 Прямое титрование
В бюретку наливают титрованный раствор трилона Б, в колбу для титрования - раствор, содержащий ион металла. Этот раствор перед титрованием доводят до определенного значения pH 9...10 (добавлением буфера, например, аммонийного). Титрование проводят в присутствии металлоиндикатора, реагирующего на изменение показателя концентрации иона металла в растворе (точнее, на изменение показателя концентрации иона металла pMe аналогично тому, как pH-индикатор реагирует на изменение pH). Раствором трилона Б титруют до тех пор, пока все катионы металла из комплекса с металлоиндикатором не перейдут в комплексонат, что сопровождается изменением окраски раствора в конечной точке.
Этим методом определяют ионы многих металлов, например, кальция, бария, стронция, никеля и др.
6.4. 2 Обратное титрование
В том случае, когда надежный индикатор на ион металла отсутствует или pH, необходимый для образования комплекса вызывает осаждение определяемого металла, применяют метод обратного титрования. Титрованный раствор трилона Б добавляют в заведомом избытке к раствору анализируемой соли. Устанавливают, вводя буферный раствор, нужное значение pH. Избыток трилона Б оттитровывают раствором хлорида магния или хлорида цинка. Точка эквивалентности фиксируется по изменению окраски индикатора (например, эриохрома черного Т). Обратное титрование применяют также, когда ионы металла взаимодействуют с титрантом или металоиндикатором медленно.
6.4 3 Титрование путем вытеснения
Этот метод применяют в тех случаях, когда:
1) определяют содержание металла, входящего в состав труднорастворимого осадка (например, MgNH4PO4);
2) нет подходящего индикатора для прямого титрования в случае блокировки индикатора (то есть индикатор связан с металлом в более прочный комплекс, чем комплекс металла с трилоном);
3) реакция идет очень медленно.
В этом случае можно соединение с комплексоном получить обменной реакцией при титровании соли определяемого металла с ЭДТА (например, раствором комплексоната магния Na2[ MgY ] или цинка Na2[ZnY]). К анализируемому раствору прибавляют комплексонат магния (его получают титрованием или используют готовый), реакция идет по уравнению:
Me2+ + Na2[ MgY ] Na2[ MeI ] + Mg2+ .
Mg2+ вытесняется в количестве, эквивалентном количеству ионов металла, связанных в прочный комплекс с ЭДТА. Вытесненный Mg2+ титруют трилоном Б.
Для применения этого метода необходимо, чтобы образующееся соединение определяемого металла с комплексоном было более прочным, чем комплексонат магния или цинка.
6.4. 4 Алкалиметрическое титрование (титрование щёлочью)
Это титрование, основанное на изменении значения pH раствора (при взаимодействии иона металла и трилона Б):
Me2+ + [ H2I ]2- [ MeI ]2- + 2H+ .
Выделение H+ строго эквивалентно содержанию металла, поэтому раствор далее титруют 0,1н раствором NaOH с индикатором метил-оранжевым или метиловым-красным.
Лекция 7 Методы осадительного титрования
7. 1 Общая характеристика метода и его классификация
Метод осадительного титрования основан на применении реакций осаждения, в результате которых образуются малорастворимые соединения. Метод осадительного титрования даёт возможность количественно определять анионы, осаждаемые катионами металлов и катионы, если титровать их анионами. Например, этот метод используют для определения таких катионов и анионов, как:
![]()
Различают следующие методы:
Аргентометрия – метод объемного анализа, основанный на применении стандартного раствора (титранта) ионов серебра. Различают три вида аргентометрии:
1)Титрование без индикатора (Гей-Люссак). В основе метода лежит реакция:
Ag++Hal ↔ AgHal↓
где Hal -ионы Cl-, J-, Br-. Суть метода заключается в том, что титрование галогенов ведут до тех пор, пока последующая порция AgNO3 не вызывает образования новых количеств осадка. В этот момент в точке эквивалентности каждая лишняя капля AgNO3 вызывает коагуляцию осадка и раствор становится прозрачным. Этот метод называется методом просветления.
2)Титрование с индикатором
Метод основан на реакции, протекающей между Ag+ и Cl- в присутствии индикатора – К2CrO4. Титруют раствор, содержащий Cl- растором, содержащим Ag+ в присутствии K2CrO4. В точке эквивалентности лишняя капля Ag+ образует с ионами CrO4- красный осадок:
![]()
Красное окрашивание осадка свидетельствует о наступлении точки эквивалентности.
3)Титрование с адсорбционным индикатором.
Меркуриметрия и меркурометрия – методы, основанные на применении в качестве титрантов растворов солей двухвалентной (меркури-ион) и одновалентной (меркуро-ион) ртути. При меркуриметрическом определении ионов хлора происходит реакция:
![]()
Точка эквивалентности определяется с помощью индикатора, образующего с избытка Hg2+ окрашенный комплекс. Этот метод применяется для определения Cl-, J-, Br-.
В основе меркурометрических определений лежит реакция:
Меркури- и меркурометрические методы имеют по сравнению с аргентометрическим методом преимущество: соли ртути менее дорогие по сравнению с серебром. Однако в то же время они более токсичны.
В связи с этим, метод осадительного титрования используют лишь тогда, когда другие химические методы не применимы. Например для определение ионов хлора, брома, серебра, бария, свинца, цинка проводят только этим методом. В таблице 1 приведены примеры осадительного титрования.
Таблица 1- Примеры осадительного титрования
Определяемый компонент |
Титрант |
Индикатор |
Br-, J-, SCN-, CO32-, CrO42-, CN-, C2O42-, PO43-, S2- |
AgNO3 |
K2CrO4 |
Br-, J-, Cl-, SeO32- |
-//- |
Fe3+ |
Zn2+ |
K4[Fe(CN)6] |
Адсорбционный |
SO42- |
Pb(NO3)2 |
Адсорбционный |
Ag+ |
NH4SNC |
Fe3+ |
Cl-, Br- |
Hg2(NO3)2 |
Адсорбционный |
Не все реакции могут быть использованы для количественного определения катионов и анионов этим методом. Основными требованиями к реакциям, применяемым в методах осадительного титрования, являются следующие:
1) Выделяющиеся осадки должны по возможности обладать минимальной растворимостью. Для метода осадительного титрования применяются такие реакции, в результате которых образуется осадок, растворимость которого не превышает 10-5моль/л
2) Реакция должна идти быстро, а посторонние вещества в растворе не должны мешать реакции.
7. 2 Кривые титрования
Процесс осадительного титрования исследуют с помощью кривых титрования, которые позволяют определить пригодность метода для анализа и подобрать индикатор. Кривые титрования изображают зависимость концентрации вещества или титранта в титруемом растворе от объёма добавленного титранта. Для удобства концентрации изображают в виде функции «р»: рА= -lgCA (по аналогии с рН). На рисунке 1 приведены кривые титрования 0,1М растворов, содержащих ионы Cl-, J- раствором нитрата серебра той же концентрации и изменение концентрации ионов серебра в процессе титрования.
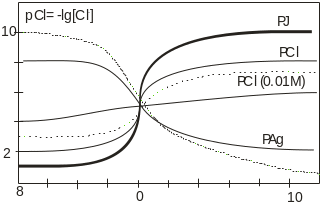
Рисунок 1- Кривые титрования ионов Cl-, J- (___) и Ag+ (---)
Таким образом, вблизи точки эквивалентности имеется резкий скачок pCl от 2 до 6 и pAg от 6 до 4. Нетрудно заметить, что величина скачка зависит от концентраций растворов. Разбавление растворов приводит к уменьшению скачка или к его исчезновению. На величину скачка титрования оказывает влияние и величина произведения растворимости осадка. Так, если титровать раствор KJ раствором AgNO3, образуется осадок AgJ (ПР = 10-16), скачок начинается при pJ 1 и заканчивается при pJ 10, т.е. занимает целых 6 единиц pJ. И, наоборот, чем больше величина произведения растворимости, тем скачок меньше (ПРAgCl= 10-10).
В связи с этим в титрометрическом анализе применяются только те реакции осаждения, при которых образующийся осадок практически нерастворим.
Метод осадительного титрования можно использовать для титрования не только индивидуальных веществ, но и для титрования смесей. Например, если в растворе имеется смесь Cl- и J- -ионов, то можно отдельно определить каждый анион. Кривая титрования будет приведена на рисунке 2.

Рисунок 2- Кривая титрования смеси ионов Cl- и J-.
Поскольку ПРAgJ = 10-16, а ПРAgCl = 10-10, то вначале оттитровываются иодид-ионы, а затем хлорид-ионы, так как сначала образуется соль AgJ, а затем, когда в растворе ионы иода уже отсутствуют, образуется соль AgCl. Для раздельного титрования ионов необходимо, чтобы произведение растворимости образующихся солей отличалось не менее, чем в 5 раз.
7. 3 Способы фиксирования точки эквивслентности в методе осадительного титрования
7. 3. 1 Безиндикаторные методы.
7. 3. 1. 1Метод равного помутнения (Гей-Люссака)
Если титровать раствор NaBr раствором AgNO3 то происходит реакции:
Br-+Ag+AgBr
Осадок выпадает до тех пор, пока в растворе есть избыток брома. Поэтому если отбирать в конце титрования небольшие порции титруемого раствора в пробирки и добавлять к ним одну каплю разбавленного раствора AgNO3, можно увидеть – есть ли помутнение раствора. Если есть, титрование ведут дальше. Осадок же AgBr коагулирует, собирается на дне сосуда в виде крупных творожных частиц. Этот метод достаточно точен но кропотлив. Поэтому на практике пользуются другими методами.
7. 3. 1. 2 Титрование до точки просветления.
Он может быть применен в том случае, если малорастворимое соединение в процессе титрования находится в коллоидном состоянии. Так, например, при титровании J- раствором Ag+ до точки эквивалентности раствор мутный, а в точке эквивалентности, когда все ионы иода оттитрованы, происходит коагуляция частиц и осаждение их в виде крупных творожных хлопьев. Раствор при этом совершенно осветляется. Этот момент называется точкой просветления.
7. 3. 2 Методы с применением индикаторов
В осадительном титровании применяют три типа индикаторов: осадительные, металлохромные (комплексообразую Cl- и J- щие) и адсорбционные.
Осадительные индикаторы образуют с титрантом цветные осадки, при появлении которых титрование заканчивают. Например, применение K2CrO4 в качестве индикатора основано на способности CrO42- образовывать с Ag+ осадок Ag2CrO4 красно-коричневого цвета. Этот осадок начинает выпадать после того, как все ионы хлора будут связаны в осадок в виде AgCl. Причина заключается в различии величины растворимости хлорида (10-10) и хромата (10-5) серебра.Первым осаждается AgCl и только после того, как все ионы хлора будут связаны в осадок AgC, первая избыточная капля Ag+ дает красно-коричневое окрашивание раствора – это появляется Ag2CrO4. Другой метод основан на применении в качестве индикатора соли железа NH4Fe(SO4)2.12H2O при титровании растворов Ag+ раствором аммония NH4SCN.
Металлохромные индикаторы дают с титрантом цветной комплекс, образующийся около точки эквивалентности. Устойчивость этого комплекса должна быть меньше, чем устойчивость осадка, получающегося при осадительном титровании так как в противном случае комплекс будет образовываться раньше осадка.
Адсорбционные индикаторы представляют собой органические соединения, являющиеся слабыми кислотами, диссоциирующими согласно уравнению
![]()
Молекулярная и ионные формы имеют разную окраску.
Анионы этих индикаторов, адсорбируясь на поверхности положительно заряженных коллоидных частиц, выпадающих в процессе титрования осадков, вызывают изменение цвета поверхности этих осадков. Если изменение цвета происходит вблизи точки эквивалентности, то можно использовать такие адсорбционные индикаторы для установления конца титрования.
Наиболее известными представителями этого класса органических соединений являются флуоресцин и эозин. Например, эозин представляет собой двунатриевую соль тетрабромфлуоресцина:
 2Na+
2Na+
Действие
этого индикатора основано на том, что,
например, при титровании NaCl
раствором AgNO3
до точки эквивалентности частицы осадка
хлорида серебра адсорбируют на всей
поверхности ионы хлора с образованием
осадка белого цвета [AgCl]m
. nCl-.
Адсорбция ионов хлора наблюдается до
тех пор, пока в растворе находятся
свободные ионы Cl-.
Поэтому, если в титруемый раствор
добавить флуоресцина или эозина
индикаторы не будут изменять окраски
до тех пор, пока в растворе имеется
избыток Cl--ионов.
Это объясняется тем, что окрашенные
анионы индикатора, несущие отрицательные
заряды, не адсорбируются отрицательно
заряженными коллоидными частицами
осадка. В момент наступления точки
эквивалентности при самом незначительном
избытке AgNO3
осадок адсорбирует избыток Ag+
и вместе с ними NO3-
ионы, при этом образуется осадок
![]() .
Ионы NO3-
не очень прочно удерживаются осадком
так как не являются с ним одноименными,
следовательно они легко замещаются на
другие анионы, способные сильнее
адсорбироваться осадком. Поэтому частицы
осадка адсорбируют интенсивно окрашенные
анионы индикатора и осадок окрашивается
в тот цвет, который присущ молекулярной
форме индикатора, что служит сигналом
к прекращению дальнейшего титрования.
.
Ионы NO3-
не очень прочно удерживаются осадком
так как не являются с ним одноименными,
следовательно они легко замещаются на
другие анионы, способные сильнее
адсорбироваться осадком. Поэтому частицы
осадка адсорбируют интенсивно окрашенные
анионы индикатора и осадок окрашивается
в тот цвет, который присущ молекулярной
форме индикатора, что служит сигналом
к прекращению дальнейшего титрования.
Лекция 8 Статическая обработка результатов анализа
1.1 Параметры, рассчитываемые при обработке результатов
В процессе анализа исследуемые
пробы подвергаются химической обработке.
Как правило, все химические реакции
считаются равновесными и несмотря на
то, что исследователи стараются выбрать
реакции, практически смещенные в сторону
продуктов, все же всегда имеют место
явления, создающие случайные и
систематические погрешности. Например,
это могут быть различные солевые эффекты,
явления соосаждения и другие, препятствующие
полноте протекания реакций.
Существует
два фактора, по которым аналитик судит
о своих результатах:
1) воспроизводимость
полученных результатов;
2) соответствие
их содержанию в пробе (правильность
результатов).
Воспроизводимость
зависит от случайной погрешности,
правильность - от систематической
погрешности.
Погрешность титриметрического
метода анализа определяется в основном
погрешностью измерения объема и
непосредственно зависит от величины
капли, объем которой составляет в среднем
0,04 см3. При тщательном титровании
можно снимать доли капли, например
половину, тогда абсолютная погрешность
измерения объема составит ± 0,02 см3,
а относительная ( при объеме, пошедшем
на титрование, 20 см3):
![]() =
0,1 %
Поэтому, чтобы не снижать
точность результатов, расчеты следует
производить с ошибкой, не превышающей
0,1 %. Для этого все численные величины
при расчетах (объемы раствора, молекулярные
массы, эквиваленты, навески и т. д.) должны
быть выражены четырьмя значащими
цифрами. Например:
1) V = 19,53, а не 19,5
см3.
2) С(H2SO4) = 0,1010, а
не 0,101 моль/дм3.
3) Т(H2SO4)
= 0,004900, а не 0,0049 г/см3.
4) 1/2М(H2SO4)
= 49,04, а не 49 г/моль.
Цель всех
аналитических исследований - нахождение
результата, наиболее близкого к истинному
содержанию в пробе. Общую погрешность
метода можно оценить только с привлечением
методов математической статистики.
Объективную оценку результата анализа
получают путем математической обработки
на основе теории ошибок. Как правило,
ни одно единичное определение не дает
истинного значения определяемой
величины. Поэтому определение проводится
несколько раз для нахождения наиболее
вероятного значения определяемой
величины. На практике при анализе всегда
имеют дело с небольшим конечным числом
измерений ( n ). Для учета влияния случайных
погрешностей на результаты анализа
рассчитывают следующие выборочные
параметры: среднеарифметический
результат определений, дисперсию,
стандартное отклонение, относительное
стандартное отклонение и
доверительный интервал.
=
0,1 %
Поэтому, чтобы не снижать
точность результатов, расчеты следует
производить с ошибкой, не превышающей
0,1 %. Для этого все численные величины
при расчетах (объемы раствора, молекулярные
массы, эквиваленты, навески и т. д.) должны
быть выражены четырьмя значащими
цифрами. Например:
1) V = 19,53, а не 19,5
см3.
2) С(H2SO4) = 0,1010, а
не 0,101 моль/дм3.
3) Т(H2SO4)
= 0,004900, а не 0,0049 г/см3.
4) 1/2М(H2SO4)
= 49,04, а не 49 г/моль.
Цель всех
аналитических исследований - нахождение
результата, наиболее близкого к истинному
содержанию в пробе. Общую погрешность
метода можно оценить только с привлечением
методов математической статистики.
Объективную оценку результата анализа
получают путем математической обработки
на основе теории ошибок. Как правило,
ни одно единичное определение не дает
истинного значения определяемой
величины. Поэтому определение проводится
несколько раз для нахождения наиболее
вероятного значения определяемой
величины. На практике при анализе всегда
имеют дело с небольшим конечным числом
измерений ( n ). Для учета влияния случайных
погрешностей на результаты анализа
рассчитывают следующие выборочные
параметры: среднеарифметический
результат определений, дисперсию,
стандартное отклонение, относительное
стандартное отклонение и
доверительный интервал.
1. Среднее значение случайной величины ( x ):
Эта величина при большом числе определений наиболее соответствует истинному значению.
x
=
![]()
![]() i
(
1 )
i
(
1 )
2. Выборочная дисперсия ( S2 ):
S2
=
 ( 2 )
( 2 )
Стандартное отклонение единичного измерения ( S ):
S
=
 (
3 )
(
3 )
Стандартное отклонение средней величины (Sх ):
Sх
=
![]() =
=
![]() ( 4
)
( 4
)
5. Относительное стандартное отклонение ( Sr ):
Sr = S /x . (5)
1. 2 Исключение сомнительных результатов
Если серия данных содержит выпадающий результат, который значительно отличается от среднего, следует решить оставить его или исключить? При исключении результатов из малой выборки следует получить дополнительные данные взамен отбрасываемых. Нужно иметь в виду, что из выборки можно исключить не более 1/3 результатов. Если же выборка содержи более 1/3 результатов, подлежащих выбраковке, то вся выборка считается некорректной.
1.2.1 S- критерий
После вычисления стандартного отклонения абсолютных величины случайных отклонения | xi -х | сопоставляют с величиной 3S. Если для всех хi выборки соблюдается условие:
xi -x 3S, ( 6 )
то все экспериментальные значения считают пригодными для расчета доверительного интервала. Если же для отдельных значений условие (6) не выполняется, то эти результаты могу быть исключены.
1. 2. 2. Q - критерий
Для исключения сомнительных результатов можно использовать Q - критерий. При проведении Q - теста все результаты определения располагают по порядку возрастания их величины и обозначают x1, x2,...xn . Для первого и последнего результатов рассчитывают:
Q1
=
![]() и Q2 =
и Q2 =
![]() .
( 7 )
.
( 7 )
Больший из этих показателей ( Q1 или Q2 ) сравнивают с табличным значением Q( Рдов,n ) для заданной доверительной вероятности и числа определений n (таблица 1). Если полученное значение Q равно пли больше приведенного в таблице 1, то сомнительный результат отбрасывают.
Если результат x1 или xn будет отброшен, то число оставшихся результатов уменьшится на 1, и тогда Q - тест повторяют для n-1 значений. Так продолжают, пока не будут отброшены все результаты, полученные с грубыми ошибками. Для оставшихся результатов рассчитывают заново значения x, S и Sx.
Таблица 1 - значения Q - теста в зависимости от числа определений ( n ) и принятой доверительной вероятности (Р).
n\ P дов |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
0,90 |
0,94 |
0,76 |
0,64 |
0,56 |
.0,51 |
.0,47 |
0,44 |
0,41 |
0,95 |
0,98 |
0,85 |
0,73 |
0,64 |
0,59 |
0,54 |
0,51 |
0,48 |
0,99 |
0,99 |
0.93 |
0,82 |
0,74 |
0,68 |
0,63 |
0,60 |
0,57
|
Q -критерий используется для выявления промахов в выборках с числом определений 3 n 10.
1. 2. 3 Доверительный интервал
При помощи статистики можно установить такие пределы области вокруг экспериментально найденного среднего х, в которых следует ожидать с данной вероятностью нахождение истинного значения. Пределы,, полученные таким образом, называются доверительными границами. Интервал, ограниченный этими пределами, называется доверительным интервалом.
Упомянутые
величины имеют строгий математический
смысл. Вероятность того, что найденное
значение измеряемой величины лежит в
интервале [
- k,
+ k
], где
и
- соответственно среднее арифметическое
и стандартное отклонение генеральной
совокупности (n
), есть доверительная вероятность.
Численно доверительная вероятность
равна
![]() ,
где h(х)
- функция Гаусса, а пределы интегрирования
обозначают доверительный интервал a=
- k,
b=+k.
Функция Гаусса описывает теоретически
ожидаемое распределение экспериментальных
результатов генеральной совокупности
Функция Гаусса имеет вид:
,
где h(х)
- функция Гаусса, а пределы интегрирования
обозначают доверительный интервал a=
- k,
b=+k.
Функция Гаусса описывает теоретически
ожидаемое распределение экспериментальных
результатов генеральной совокупности
Функция Гаусса имеет вид:
![]() ,
(8)
,
(8)
где x- единичное измерение, n - частота появления каждого значения х.
Для абсолютно правильного предсказания мы должны были бы выбрать достаточно большой интервал и включить в него все мыслимые значения, которые может принимать xi. Для аналитических определений такой интервал не имеет ценности. Если мы примем, что вероятность попадания в интервал составляют 99 правильных результатов из 100, он не будет столь большим. Его можно сделать еще меньше, если считать приемлемой вероятность 0,95.
Для ограниченного числа измерений доверительные границ средней величины рассчитывают, используя статистический критерий Стьюдента t=F(Pдов,f). В аналитической химии обычно принимают Рдов=0,95. Константу t для избранной доверительной вероятности и числа степеней свободы f= n-1, часто называемую коэффициентом Стьюдента, находят по таблице 2.
Таблица 2 - Значения коэффициентов Стьюдента
f \Pдов |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
0,95 |
12,7 |
4,30 |
3,18 |
2,78 |
2,57 |
2,45 |
2,37 |
2,31 |
2,26 |
2,23 |
0,99 |
63,6 |
9,93 |
5,84 |
4,60 |
4,03 |
3,71 |
3,59 |
3,36 |
3,25 |
3,17 |
С использованием коэффициента Стьюдента доверительный интервал выселяют как:
t.Sx
или t.S/![]() .
.
Таким образом, результат анализа представляют в :
x t.S/ .
1. 2. 3 Значащие цифры
Результат анализа должен быть выражен числом, которое содержит только значащие цифры. Значащие цифры - это все достоверно известные цифры данного числа плюс первая недостоверная цифра.
Для оценки достоверности результатов аналитических определений следует учитывать реальные возможности используемого метода или методики. В качестве статистических критериев при этом может служить, например, стандартное отклонение или доверительный интервал. При отсутствии конкретных данных считают, что недостоверность последней цифры равна ± 1.
Нуль, стояний в середине или в конце числа, является значащей цифрой. При правильной записи результата анализа нули, которые не являются значимыми, следует исключить, а результат анализа представить в виде числа, содержащего произведение значащих цифр на 10n.Например, если число 1250 содержит три значащие цифры, то правильно его представить в виде 1,25-103, а если в этом же числе четыре значаще цифры, то следует записать его как 1,250-103.
Нули, стоящие в начале числа, не являются значащими, они лишь показывают место запятой в десятичной дроби. Например, значение массы образца 0,03750г можно представить в виде 3,750-10-2г, если взвешивание проводили на микровесах с точностью до ± 0,00002 г; а если взвешивание проводил на аналитических весах с точностью до ± 0,0002 г, то правильное представление результата: 0,0375 г или 3,75-10-2 г.
Незначащие цифры исключают, округляя число. При этом, если отбрасываемая цифра меньше 5, последняя значащая цифра не изменяется. Если отбрасываемая цифра больше или равна 5, последняя значащая цифра увеличивается на единицу. Если за первой недостоверной цифрой следует цифра 5, то применяют другое правило: округляют цифру 5 до ближайшего четного числа.
Количество значащих цифр в числе, полученном в результате всех вычислений, определяется из сравнения недостоверности чисел. При сложении и вычитании количество знаков обусловлено числом, которое имеет наибольшую абсолютную недостоверность и наименьшее количество десятичных знаков. При умножении и делении количество значащих цифр обусловлено числом, имеющим наибольшую относительную недостоверность. Логарифмические величины должны содержать такое же количество значащих цифр (после запятой), как и использованные при расчете нестепенные числа. Например, рН раствора с концентрацией [Н+]= 1,7.10-4 М равен 0,77. Это объясняется тем, что цифры, полученные при логарифмировании степенного члена, не являются значащими.
В полумикроанализе (гравиметрия, титриметрия) результат должен быть представлен в виде числа, содержащего четыре значащие цифры, что связано с точностью измерений массы веществ и объемов растворов.
Пример расчета №1 Данные определения основного компонента в пробе лекарственного препарата представлены в таблице. Определить его истинное значение и рассчитать доверительный интервал.
№ |
Х(%) |
Хi
- |
(Х - )2 |
1 |
96,6 |
+0,6 |
0,36 |
2 |
95,4 |
-0,6 |
0,36 |
3 |
95,5 |
-0,5 |
0,25 |
4 |
96,5 |
+0,5 |
0,25 |
5 |
96,1 |
+0,1 |
0,01 |
6 |
95,9 |
-0,1 |
0,01 |
7 |
94,1 |
|
|
![]() Проверим на промах величину, наиболее
отличающуюся от результатов всей серии-
94,1%.
Проверим на промах величину, наиболее
отличающуюся от результатов всей серии-
94,1%.
![]() Табличные
значения Q для n = 7 и Р = 95 % - 0,480, следовательно
это значение является промахом и не
включается в расчет среднего арифметического
значения.
Табличные
значения Q для n = 7 и Р = 95 % - 0,480, следовательно
это значение является промахом и не
включается в расчет среднего арифметического
значения.
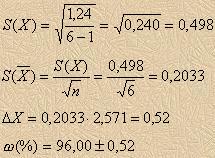
Пример №2
При определении массовой доли фосфорной кислоты в образце были получены результаты, приведённые в таблице. Рассчитать абсолютную и относительную ошибку опытов.
№ определения |
Содержание
|
|
|
S2, S |
1 |
0,7594 |
+0,0010 |
0,00000100 |
|
2 |
0,7543 |
-0,0041 |
0,00001681 |
|
3 |
0,7623 |
+0,0039 |
0,00001521 |
|
4 |
0,7594 |
+0,0010 |
0,00000100 |
|
5 |
0,7605 |
+0,0021 |
0,00000441 |
|
6 |
0,7543 |
-0,0041 |
0,00001681 |
|
|
|
|
|
|

Стандартное отклонение среднего:
![]() ;
;
Принимая
надежность
![]() по таблицам для
по таблицам для
![]() ,
т.е.
,
т.е.
![]() находим
находим
![]() .
.
Тогда доверительный интервал:

Абсолютная
ошибка:
![]() .
.
В заключении высчитывают относительную ошибку в процентах по формуле:
![]() ;
;
![]() .
.
Все результаты аналитической обработки сводятся в таблицу.
-
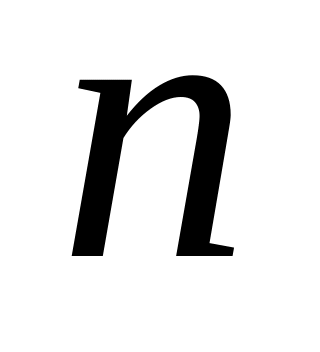





6
0,7584
0,00001104
0,0033
0,0036
0,45
Литература
Основная
1. Основы аналитической химии. /Под ред. Ю. А. Золотова. -Кн. 1-2.-М.: Химия, 1999.
2. Кунце У., Шведт Г. Основы качественного и количественного анализа. – М. : Мир,1997. - 424с.
3. Аналитическая химия. Кн.1. Титриметрические и гравиметрические методы анализа. – М. : Дрофа, 2002.- 368с.
4. Алексеев В. Н. Количественный анализ. – М.: Химия, 1972 . –504с.
5. Скуг Д. А., Уэст Д. М. Основы аналитической химии. -М. : Мир, 1979. - 480с.
6. Крешков А. П. Основы аналитической химии.- М.: Химия,1976. В 2т. –1123с.
Дополнительная
6. Петерс Д., Хаме Дж., Хифтье Г. Химическое разделение и измерение -. М :Химия. 1978. -198с.
7. Пикеринг У. Современная аналитическая химия.- М.: Мир, 1977. -229с.
8. Янсон Э.Ю., Петпинь Я. К. Теоретические основы аналитической химии.- М. :Высшая школа,1980. -452с.
9. Пономарёв В. Д. Аналитическая химия.- М. : Высшая школа, 1982. - 304с.
10. Бончев П.Р. Введение в аналитическую химию.- Л. : Высшая школа, 1982. 495с.
11. Шарло Г. Методы аналитической химии. -М. : Химия, 1978. -975с.
12. Задачник по количественному анализу под ред. Мусакина А. П. -Л. : Химия. -1972. -376с.
13. Лурье Ю.Ю. Справочник по аналитической химии. -М. : Химия. 1979. -480с. 1экз.
14. Справочник химика-аналитика. - М. : Металлургия. 1976. -257с. 3 экз.
15. Доерфель К. Статистика в аналитической химии.- М.: Мир.1969. -196с. 2 экз.
16. Чарыков А. К. Математическая обработка результатов химического анализа. Методы обнаружения и оценки ошибок. -М. : Химия. 1984. -254с. 1 экз.