

Электрофильные реакции с олефинами и ароматическими двойными связями
Реакционная способность двойной связи
Двойная связь С=С вследствие наличия легкодоступных (поляризуемых) -электронов представляет собой основание Льюиса и соответственно вступает в реакции с кислотами или кислотами Льюиса. В результате возникают либо рыхлые аддукты (-комплексы) 7.1 или 7.2., либо карбкатионы (-комплексы) 7.3.
В -комплексах -связь в направлении своей наибольшей протяженности (перпендикулярно плоскости расположения заместителей) перекрывается с s или р-орбиталью кислоты (1 на схеме 7.1.). Поскольку двойная связь выступает в роли донора электронов, а кислота – в роли акцептора электронов, то можно также сказать о донорно-акцепторных соединениях или комплексах с переносом заряда.
-Комплексы образуются также с акцепторами, которые у своего центрального атома не имеют низко расположенных d-орбиталей, например, с галогеноводородами или другими протонными кислотами. Если же центральный атом кислоты имеет низкорасположенные, не занятые в исходном сотоянии d-орбитали, то эти орбитали при переносе заряда частично заполняются электронами, которые могут быть далее перенесены на возбужденную (антисвязывающую) -орбиталь олефина (см.2 на схеме 7.2.)

У ароматических соединений помимо локализованных -комплексов (4 на схеме 7.1) существуют также комплексы типа 3 на схеме 7.2 прежде всего с ионами или нуль-валентными состояниями «мягких» переходных металлов – ртути, серебра, железа, кобальта, никеля, платины, палладия, родия, молибдена.
В комплексах аренов с хинонами, нитроаренами или тетрацианэтиленом и донор, и акцептор имеют дополнительные -связи, поэтому возможно и дополнительное -–перекрывание, когда партнеры распологаются друг над другом, как показано на схеме 7.2, 3.
Комплексы переходных металлов с олефинами и ацетиленами приобрели большое значение в органическом синтезе, например, при гомогенном гидрировании, димеризации и олигомеризации олефинов и диенов, прямом окислении олефинов, карбонилировании и гидроформилировании.
В -комплексах типа 7.1. акцептор X-Y может быть либо только поляризованным (7.1., 3), либо ионизованным (7.1., 2). Созданию ионизированной формы благоприятствуют высокая основность С=С-связи и большая сольватирующая способность среды.
Между -комплексами и -комплексами возможны взаимные переходы. -Комплексы образуются тогда, когда анион Y кислоты X-Y особенно энергетически выгоден. Так, из аренов и безводного фтористого водорода образуются только -комплексы, из тех же компонентов в присутствии трехфтористого бора – -комплексы (карбкатионы).
Для аренов, где известны как - так и -комплексы, установлено, что зависимость констант образования -комплексов от заместителя пропорциональна соответствующей зависимости для -комплексов; следовательно, и -комплексы можно использовать для определения относительной основности. Однако вследствие рыхлой связи константы устойчивости -комплексов значительно меньше зависят от природы заместителей у С=С-связи, чем это наблюдается у -комплексов. Поэтому такие константы не являются хорошим измерителем реакционной способности олефинов и аренов.
Из констант образования комплексов с I2 и SO2 получен следующий ряд повышения основности, находящийся в согласии с ожидаемым на основании индукционных и мезомерных эффектов заместителей при двойной связи:
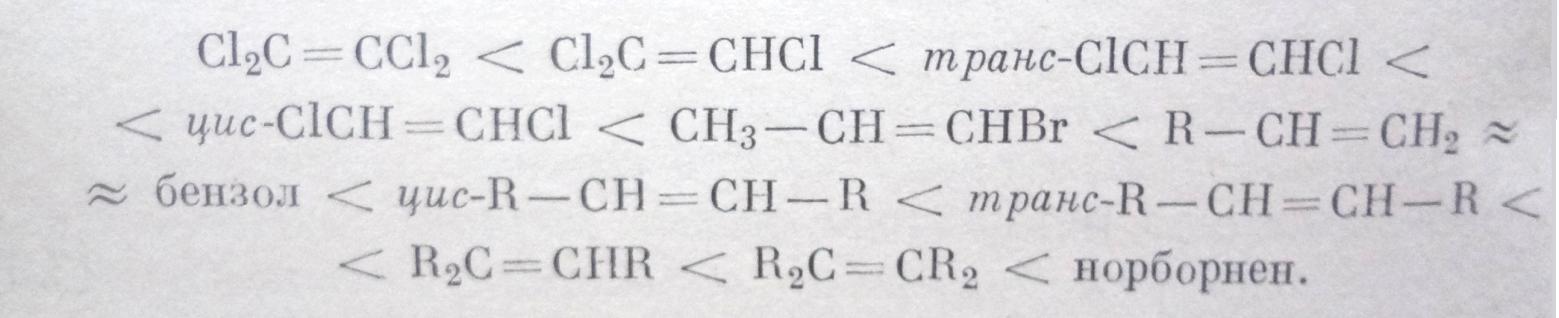
Таким образом, олефины в общем более основны, чем бензол.
Известно, что кислоты легко присоединяются к олефинами, в то время как основания обычно не вступают в реакцию. Поскольку эти реакции проходят в условиях, исключающих радикальные процессы, то принимают ионный механизм, и соответственно, скорость реакции обнаруживает хорошую пропорциональность с основностью олефинов.
Можно дать следующее обобщенное изображение реакций ионного присоединения к олефинам:
 7.4
7.4
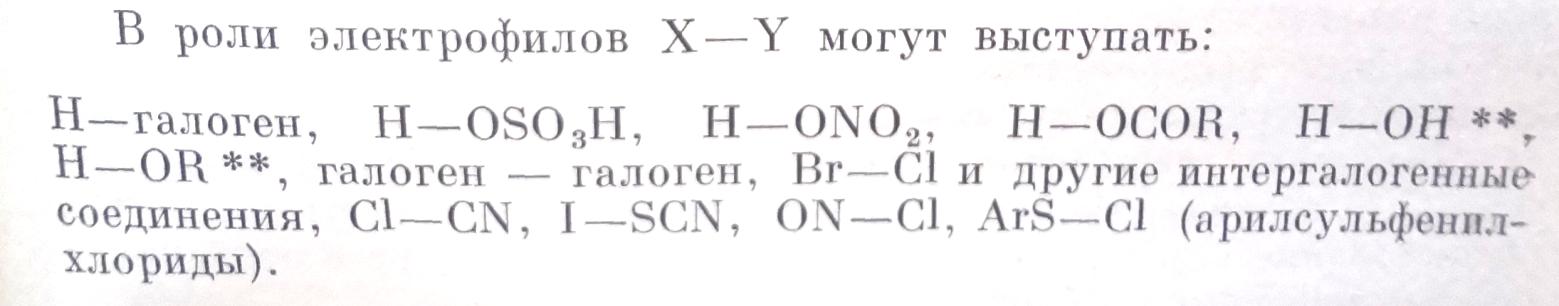
Так как речь идет об электрофильном присоединении X-Y, то все реакции присоединения к различным образом замещенным олефинами имеют отрицательные гамметовские константы реакции (благоприятствуют электродонорные группы в олефинах).
Карбкатион 3, (схема 7.4.), образующийся в качестве промежуточного продукта, имеет свою собственную судьбу: он может присоединять нуклеофил Y(из реагента X-Y) либо другие присутствующие нуклеофилы, например растворитель ROH, может перегруппировываться, может отщепить протон с образованием олефина, например:
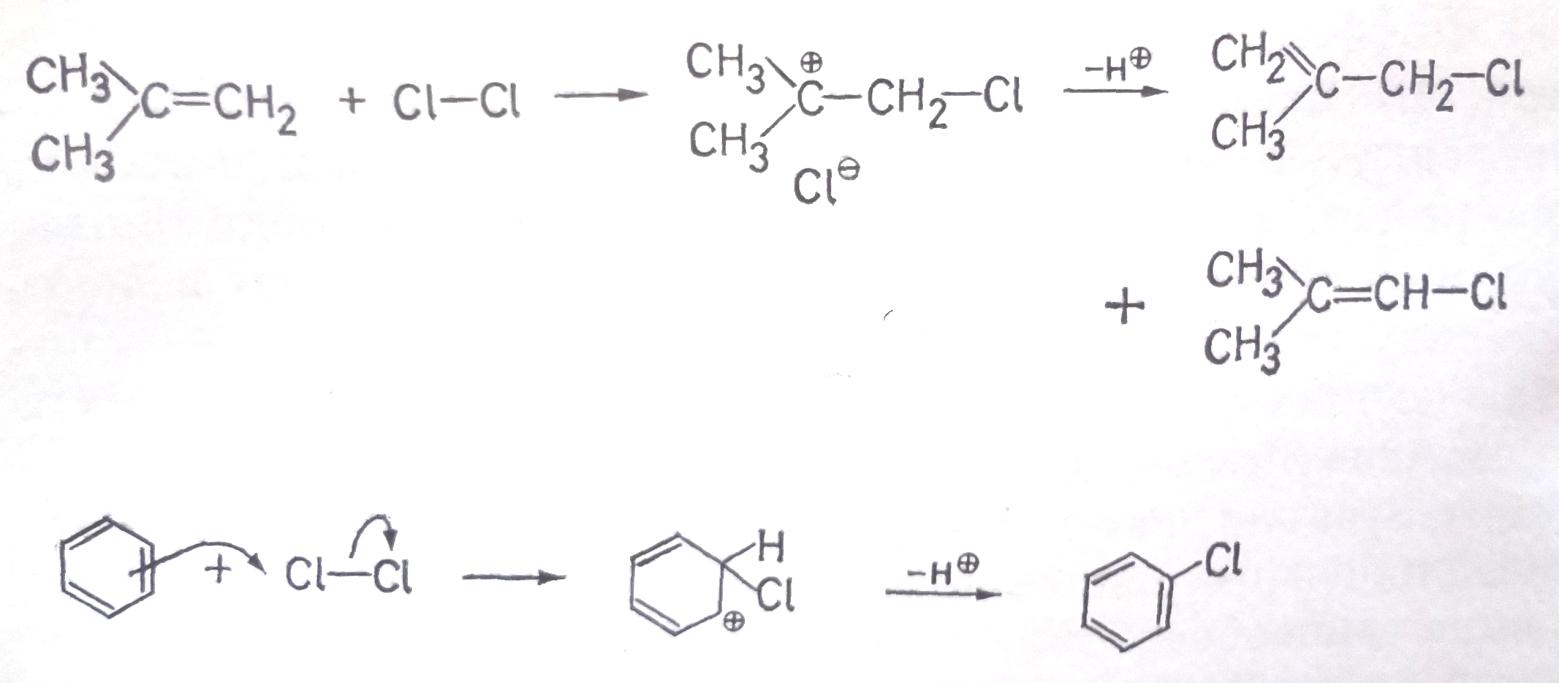
В последнем случае в итоге происходит реакция замещения (зависит от термодинамической устойчивости, запаса энергии в конечных продуктах).
Поскольку скорость взаимодействия электрофильных реагентов с двойными связями явно зависит от силы кислотно-основного взаимодействия, значительное влияние оказывает и реагент. Чем выше его кислотность, тем легче должен образовываться из двойной связи -комплекс и карбкатион. Например, сила галогенов, как кислот Льюиса возрастает с ростом их электроотрицательности, так что наибольшей реакционной способностью обладает фтор.
Кислотность всех галогенов можно повысить действием известных «переносчиков галогена», как тригалогениды алюминия, железа или другие сильные кислоты Льюиса. Эти катализаторы, в частности, находят применение при реакциях замещения в аренах. Их действие основано на электроноакцепторных свойствах

Как и у галогенов, реакционная способность протонных кислот также растет с повышением их кислотности, например в ряду:
![]()