

Денатурация, причины и признаки, использование в медицине.
Белки чувствительны к внешним воздействиям. Нарушение пространственной структуры белков называют денатурацией. При этом белок теряет все свои биологические и физико-химические свойства. Денатурация сопровождается разрывом связей, стабилизирующих "нативную" структуру белка. Как уже отмечалось выше, в основе стабилизации структуры белков основную роль играет слабое взаимодействие, поэтому денатурацию могут вызывать различные факторы: нагревание, облучение, механическое встряхивание, охлаждение, химическое воздействие. При денатурации, как правило, нарушается и растворимость белков, так как нарушение структуры приводит к появлению на поверхности большого числа гидрофобных групп, обычно упрятанных в центре белковой молекулы.
Первичная структура белка при денатурации не изменяется, что позволило показать возможность восстановления функций и структуры денатурированного белка, хотя в большинстве случаев денатурация является необратимым процессом. В лабораторной практике денатурация используется для депротеинизации биологических жидкостей. Факторы, вызывающие денатурацию, называют денатурирующими агентами. К ним можно отнести:
1. Нагревание и действие облучения высоких энергий (ультрафиолетовое, рентгеновское, нейтронное и т.д). В основе лежит возбуждение колебаний атомов, сопровождающееся разрывом связей.
2. Действие кислот и щелочей; изменяют диссоциацию групп, уменьшают число ионных связей.
3. Ионы тяжелых металлов. Образуют комплексные соединения с группами белка, что сопровождается разрывом слабого взаимодействия.
4. Восстановители - вызывают разрыв дисульфидных мостиков.
5. Мочевина, гуанидиний хлористый - формируют новые водородные связи и разрывают старые. Явление денатурации можно использовать и для качественного анализа присутствия белков в растворах. Для этого пользуются пробой с кипячением исследуемой жидкости после ее подкисления. Образующееся при этом помутнение связано с денатурацией белка. Часто используют и осаждение органическими кислотами: сульфосалициловой или трихлоруксусной.
Ферменты. Особенности ферментативного катализа. Строение и структура ферментов.
Краткая история энзимологии.
Присуждением Нобелевской премии Дж Самнеру, Дж. Нортропу и Стенли в 1946 г была подведена черта длительному периоду развития энзимологии – науки о ферментах. Начало этой науки восходит к заре истории развития человечества, использующего ряд технологических ферментативных процессов в своей жизни: хлебопечение, виноделие, обработка шкур животных и т.д. Потребность совершенствования этих процессов стало побудительным началом для их углубленного исследования. К первым научным описаниям ферментативных процессов относится описание пищеварения у животных Рене Антуан реомюр (1683—1757) при постановке своих экспериментов исходил из сделанного Фолкнером предположения о том, что хищные птицы отрыгивают не переваренные остатки пищи. Реомюр сконструировал маленькую проволочную капсулу, в которую был положен кусок мяса и дал ее склевать сарычу. Через 24 часа птица выплюнула эту капсулу. В ней остался размягченный кусок пиши, который однако не портился. «Этот процесс может быть только результатом действия какого-то растворителя»,— заключил Реомюр. Лаззаро Спалланцани (1729-1799), профессор истории естествознания в Университете города Падуя, сообщал о подобных же экспериментах. Однако он не рассматривал пищеварение как процесс ферментации по той простой причине, что при этом не образовывались пузырьки газа.
Позже процесс ферментации был более подробно изучен одним из основоположников современной химии Антуаном Лораном Лавуазье (1743-1794). Изучая спиртовое брожение, происходящее при изготовлении вина, он обнаружил, что глюкоза превращается в спирт и углекислый газ,
К началу XIX в. преобладала общая точка зрения, что ферментация - это химические изменения, вызываемые некоторыми специальными формами органического материала, а именно «ферментами». В 1814 г. русский ученый (немец по происхождению) академик Петербургской Академии наук Константин Готлиб Сигизмунд Кирхгоф (1764-1833) показал, что образование сахара из крахмала в проросших зернах злаков обусловлено химическим процессом, а не появлением ростков. В 1810 г Ю. Гей-Люссак выделил основные конечные продукты жизнедеятельности дрожжей – спирт и углекислый газ. Я. Берцелиус, один из основоположников теории химического катализа и автор самого термина «катализ» в 1835 году подтверждает эти данные, отметив, что диастаза (экстракт из солода) катализирует гидролиз крахмала более эффективно, чем минеральная серная кислота. Важную роль в развитии энзимологии сыграл спор Ю Либиха с известным микробиологом Л. Пастером, который считал, что процессы ферментации могут происходить только в целой живой клетке. Ю. Либих, напротив, считал, что биологические процессы вызываются действием химических веществ, которые в последствии были названы ферментами. Термин энзим ( греч. еn– в,zyme- дрожжи) предложил 1878 г Фридрих Вильгельм Кюне чтобы подчеркнуть, что процесс идет в дрожжах в противоположность самим дрожжам, которые катализируют процесс ферментации. Однако в 1897 году Э. Бюхнер получил свободный от клеток экстракт из дрожжей, способный получать этанол и утвердил мнение Либиха.
Попытки объяснить одно из важных свойств ферментов специфичность привело в 1894 году немецкого химика и биохимика Э. Фишера к предложению модели взаимодействия фермента и субстрата, названной «ключ-замок» – геометрической комплементарности форм субстрата (ключ) и фермента(замок). В 1926 году Дж. Самнер после почти 9-летених исследований доказал белковую природу фермента уреазы. В те же годы Дж Нортроп и М Кунитц указали на прямую корреляцию между активностью кристаллических пепсина, трипсина и количеством белка в исследуемых образцах, приведя тем самым весомые доказательства белковой природы ферментов, хотя окончательные доказательства были получены после определение первичной структуры и искусственного синтеза ряда ферментов. Основные представления о ферментах получены уже во второй половине ХХ столетия. В 1963 году исследована аминокислотная последовательность РНКазы из поджелудочной железы. В 1965 г показана пространственная структура лизоцима. За последующие годы очищены тысячи ферментов и получено много новых данных о механизмах действия ферментов, их пространственной структуре, регуляции реакций, катализируемых ферментами. Обнаружена каталитическая активность у РНК (рибозимы). Получены антитела с ферментативной активностью –абзимы. Эта глава кратко знакомит с современными представлениями о строении, механизме действия и медицинских аспектах энзимологии.
Особенности ферментативного катализа.
1. Белковая природа катализатора
2. Исключительно высокая эффективность. Эффективность биологического катализа превышает эффективность неорганического в 109- 1012
3. Исключительно высокая специфичность:
а) абсолютная, когда фермент работает только со своим субстратом (фумараза с транс-изомерами фумаровой кислоты и не будет с цис-изомерами);
б) групповая - специфичен для узкой группы родственнных субстратов (ферменты ЖКТ).
4. Работает в мягких условиях (t=37, рН 7.0, определенные осмолярность и солевой состав).
5. Многоуровневая регуляция: регуляция активности на уровне условий среды, на уровне метаболона, на генетическом уровне, тканевом, клеточном, с помощью гормонов и медиаторов, а также с помощью субстратов и продуктов той реакции, которую они катализируют.
6. Кооперативность: ферменты способны организовывать ассоциации - продукт 1-го фермента, является субстратом для 2-го; продукт 2-го - субстратом для 3-го и т.д.
Кроме того, ферменты обладают адаптивностью, т. е. могут изменять свою активность и образовывать новые ассоциации.
7. Способны катализировать как прямую так и обратную реакцию. Направление реакции для многих ферментов определяется соотношением действующих масс.
8. Катализ жестко расписан, т. е. происходит поэтапно.
Специфичность действия ферментов.
Высокая специфичность ферментов обусловлена, конформационной и электростатической комплементарностью между молекулами субстрата и фермента и уникальной структурой активного центра фермента, обеспечивающими «узнавание», высокое сродство и избирательность протекания одной какой-либо реакции.
В зависимости от механизма действия различают ферменты с относительной или групповой специфичностью и с абсолютной специфичностью.
Для действия некоторых гидролитических ферментов наибольшее значение имеет тип химической связи в молекуле субстрата. Так например, пепсин, расщепляет белки животного и растительного происхождения, хотя они могут отличаться по химическому строению, а/к составу, физиологическим свойствам. Однако пепсин не расщепляет углеводы и жиры. Это объясняется тем, что местом действия пепсина является пептидная связь. Для действия липазы таким местом является сложно-эфирная связь жиров.
Т. е. эти ферменты обладают относительной специфичностью.
Абсолютной специфичностью действия называют, способность фермента катализировать превращение только единственного субстрата и любые изменения в структуре субстрата делают его недоступным для действия фермента. Например: аргиназа, расщепляющая аргинин; уреаза, катализирующая распад мочевины.
Имеются доказательства существования стереохимической специфичности, обусловленной существованием оптически изомерных L- и D- форм или геометрических (цис- и транс-) изомеров
Так известны оксидазы L и D а/к.
Если какое-либо соединение существует в форме цис- и трансизомеров, то для каждой из этих форм, существует свой фермент. Например, фумараза катализирует превращение только фумаровой кислоты (транс-), но не действует на цис-изомер - малеиновую кислоту.
Строение фермента.
Ферменты как и белки делятся на две группы: простые и сложные.
Простые полностью и целиком состоят из а/к и при гидролизе образуют исключительно а/к. Их пространственная организация ограничена третичной структурой. Это в основном ферменты ЖКТ: пепсин, трипсин, лизоцим, фосфатаза.
Сложные ферменты кроме белковой части содержат и небелковый компонент (кофактор).
Эти небелковые компоненты отличаются по прочности связывания с белковой частью (апоферментом).
Если константа диссоциации сложного фермента настолько мала, что в растворе все ПП цепи оказываются связанными со своими небелковыми компонентами и не разделяются при выделении и очистке, то небелковый компонент называется простетической группой и рассматривается как интегральная часть молекулы фермента.
А под коферментом понимают дополнительную группу, легко отделяемую от апофермента, при диссоциации.
Между апоферментом и простетической группой существует ковалентная связь, довольно прочная.
Между апоферментом и коферментом существуют нековалентные связи (водородные или электростатического взаимодействия). Типичными представителями коферментов являются В1 (тиамин) - пирофосфат (он содержит В1); В2 (рибофлавин) - ФАД, ФМН (ОВР); РР - НАД, НАДФ (ОВР); Н (биотин) - биоцитин (карбоксилирование); В6 (пиридоксин) - пиридоксальфосфат; пантотеновая кислота - коэнзим А.
Многие двухвалентные металлы (Сu2+, Fe2+, Fe3+, Mn2+, Мg2+, Ca2+) тоже выполняют роль кофакторов, хотя и не относятся ни к простетическим группам.
Металлы входят в состав активного центра или стабилизируют оптимальный вариант структуры активного центра.
|
Металл |
Фермент |
|
Fe2+, Fe3+ |
гемоглобин, каталаза, пероксидаза |
|
Cu+, Cu2+ |
цитохромоксидаза |
|
Zn2+ |
ДНК-полимераза, алкоголь-дегидрогеназа |
|
Mg2+ |
гексокиназа |
|
Mn2+ |
аргиназа |
|
Ni2+ |
уреаза |
|
Se2+ |
глутатионредуктаза |
Кофакторную функцию могут выполнять также: HS-глутатион, АТФ, липоевая кислота, т-РНК.
Следует отметить одну отличительную особенность двухкомпонентных ферментов, заключающуюся в том, что ни кофактор (кофермент или простетическая группа) ни апофермент в отдельности каталитической активностью не обладают и только их объединение в единое целое, протекающее в соответствии с программой их трехмерной организации, обеспечивает быстрое протекание химических реакций.
Полиферментные комплексы, метаболоны.
Метаболон – мультиферментный комплекс, состоящий из белков-ферментов обладающих всеми уровнями структурной организации и катализирующих отдельные метаболические пути. Эти комплексы обычно связаны с клеточными мембранами, играют важную роль в эволюции живых систем, поскольку обеспечивают высокую скорость катализа, тонкую, точную регуляцию и направленность (векторность) метаболизма во времени и пространстве.
В промежуточном метаболизме имеются мультиферментные комплексы, катализирующие сложную многостадийную реакцию окислительного декарбоксилирования 2-кетокислот и переноса образующегося ацильного остатка на кофермент А. В качестве акцептора электронов выступает НАД+. Кроме того, в реакции участвуют тиаминдифосфат, липоамид и ФАД. К дегидрогеназам кетокислот относятся: а) пируватдегидрогеназный комплекс (ПДГ, пируват→ацетил-КоА), б) 2-оксоглутаратдегидротеназный комплекс цитратного цикла (ОГД, 2-оксоглутарат→сукцинил-КоА) и в) участвующий в катаболизме разветвленных цепей валина, лейцина и изолейцина дегидрогеназный комплекс. Примером является ПДГ-комплекс.
А. Пируватдегидрогеназа: реакция
В пируватдегидрогеназной реакции участвуют три различных фермента [1-3]. Пируватдегидрогеназа (Е1) катализирует декарбоксилирование пирувата, перенос образованного гидроксиэтильного остатка на тиаминдифосфат(TPP, 1а), а также окисление гидроксиэтильной группы с образованием ацетильного остатка. Этот остаток и полученные восстановительные эквиваленты переносятся на липоамид (1б). Следующий фермент,дигидролипоамидацетилтрансфераза (Е2) переносит ацетильный остаток с липоамида накофермент А(2), при этом липоамид восстанавливается до дигидролипоамида. Последний снова окисляется до липоамида третьим ферментом,дигидролипоамиддегидрогеназой (Е3) с образованием НАДН + Н+(NADH + Н+) (3). Электроны переносятся на растворимый НАД+черезФАДи каталитически активный дисульфидный мостик субъединицы Е3.
Пять разных коферментовэтой реакции различными способами ассоциированы с белковыми компонентами ферментов. Тиаминдифосфат нековалентно связан на Е1, Липоамид ковалентно связан с остатком лизина Е2, а ФАД прочно ассоциирован в видепростетической группы на Е3. НАД+(NAD+) и кофермент А взаимодействуют с комплексом в виде растворимых коферментов.
Б. Пируватдегидрогеназный комплекс Escherichia coli
Пируватдегидрогеназный комплекс (ПДГ-комплекс) бактерии Escherichia coli достаточно подробно исследован. Он имеет молекулярную массу от 5,3 · 106Да и диаметр больше 30 нм, т. е. ПДГ-комплекс крупнее, чем рибосома. Комплекс состоит из 60 полипептидов (1,2): 24 молекулы Е2 (8 тримеров) образуют ядро комплекса кубической формы. Каждая из 6 граней этого куба занята димерами (возможно, тетрамерами) компонентов Е3, в то время как на 12 ребрах куба лежат димеры молекул Е1. Другие дегидрогеназы кетокислот построены аналогично, но могут отличаться числом субъединиц и молекулярной массой.

Пространственная организация составных частей комплекса очень важна для катализа. Кофермент, липоевая кислота, очень подвижен благодаря образованию связи с лизиновым остатком фермента Е2. "Ручка" липоамидадлиной примерно 1,4 нм в процессе катализа перемещается между E1 и Е3 (3). Липоамид таким способом может взаимодействовать как со связанным в Е1 тиаминдифосфатом, так и с растворимым коферментом А и акцептирующим электроны ФАД в Е3. Белковый домен ацетилтрансферазы, который связывает липоевую кислоту, очень гибок. Это дополнительно повышает дальность действия липоамидной «ручки».
|
Заведующий кафедрой биологической химии, д.м.н., проф. |
Грицук А. И. |
___________ |
21.10.2006
Министерство здравоохранения Республики Беларусь
УО «Гомельский государственный медицинский университет»
Кафедра биологической химии
Обсуждено на заседании кафедры (МК или ЦУНМС)
Протокол № _________________200__года
ЛЕКЦИЯ по биологической химии
наименование дисциплины
для студентов _2__ курсалечебногофакультета
Тема Ферменты 2. Механизм действия.
Время 90 мин.
Учебные и воспитательные цели:
Дать представление:
О механизме действия ферментов. Об этапах ферментативного катализа.
О факторах, определяющих активность ферментов [E], [S], [P], Km. О влиянии pH, давления, температуры, ионной силы на активность ферментов.
Об изостерической и аллостерической регуляция.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф. Биологическая химия. М.: Медицина, 1990. С. 105–123; 1998. С. 143–156.
Николаев А. Я. Биологическая химия. М.: Высшая школа, 1989. С. 52–92.
Дополнительная
Марри Р. и др. Биохимия человека. М.: Мир, 1993. Т. 1. С. 76–110.
Филиппович Ю. Б. Основы биохимии. М.: Высшая школа, 1993. С. 100–111.
Ленинджер А. Л. Основы биохимии. М.: Мир, 1985. Т. 1. С. 226–302.
Албертс Б. и др. Молекулярная биология клетки. М.: Мир, 1994. Т. 1. С. 113–171.
МАТЕРИАЛЬНОЕ ОБЕСПЕЧЕНИЕ
1. Мультимедийная презентация.
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№ п/п |
Перечень учебных вопросов |
Количество выделяемого времени в минутах |
|
|
Механизм действия ферментов. Этапы ферментативного катализа. |
30 |
|
|
Факторы, определяющие активность ферментов [E], [S], [P], Km. Влияние pH, [P], tº, ионной силы на активность ферментов. |
40 |
|
|
Изостерическая и аллостерическая регуляция. |
20 |
Всего 90 мин
Механизм действия ферментов. Этапы ферментативного катализа.
После установления химической природы фермента было подтверждено представление Михаэлиса и Ментен о том, что при энзиматическом катализе фермент соединяется с субстратом (они подходят как ключ к замку), образуя нестойкий промежуточный фермент-субстратный комплекс, который в конце реакции распадается с освобождением фермента и продукта реакции.
Даниэль Кошланд предложил теорию «индуцированного» соответствия: т. е. что субстрат навязывает активному центру свою форму, а активный центр в свою очередь подгоняет форму субстрата под свою собственную.
Выдвинутая в 1913 году Л. Михаэлисом и М. Ментен общая теория ферментативного катализа постулировала, что фермент Е сначала обратимо и относительно быстро связывается с со своим субстратом S в реакции:
E + S == ES
Образовавшийся при этом фермент-субстратный комплекс ES, не имеющий аналогий в органической химии и химическом катализе, затем распадается в второй более медленной (лимитирующей) стадии реакции:
ES == Е + Р
1 этап: происходит сближение и ориентация субстрата относительно субстратного центра фермента и его постепенное «причаливание» к «якорной» площадке.
2 этап: напряжение и деформация: индуцированное соответствие - происходит присоединение субстрата, которое вызывает конформационные изменения в молекуле фермента приводящие к напряжению структуры активного центра и деформации связанного субстрата.
3 этап: непосредственный катализ.
В основе химических реакций лежит образование и разрыв химических связей
Все химические реакции сопровождаются, как правило, разрывом или образованием ковалентных связей. По характеру разрыва ковалентных связей различают три типа реакций
1. Гетеролитический разрыв связи: ковалентная связь разрывается таким образом, что электронная пара остается с одним из атомов, образующим связь. Атом при этом приобретает отрицательный заряд, а у второго атома возникает положительный заряд. Такие реакции называют также ионными. Гетеролитически разрываются полярные или легкополяризующиеся ковалентные связи. Реакция катализируется кислотами или основаниями;
Гомолитический разрыв связи: при разрыве ковалентной связи электронная пара разделяется между атомами с образованием свободных радикалов. Свободные радикалы - это атомы или группы атомов с неспаренным валентным электроном.
Согласованные реакцииотличаются от 1 и 2 тем, что разрыв старых связей и образование новых происходит одновременно без образования радикалов или ионов
2. Нуклеофильные реагенты:богатые электронами соединения: а) отрицательно заряженные ионы, б) нейтральные молекулы, имеющие свободную неподеленную пару электронов, в) реагенты, способные давать карбанионы в ходе реакций. Нуклеофилы реагируют с органическими субстратами по их положительному реакционному центру. Легко образуют ковалентные связи. Биологически важными нуклеофилами являются аминогруппы, гидроксильные группы, имидазольные группы и сульфгидрильные группы аминокислот. Нуклеофильные формы этих групп одновременно являются основаниями. Связываясь с Н+,- они основания, реагируя с другими электрондефицитными центрами – они нуклеофилы.
Электрофильные реагенты : Электрондефицитные соединения: а) положительно заряженные ионы; б) нейтральные молекулы с частично положительным зарядом на одном из атомов. Электрофилы реагируют с органическими субстратами по их отрицательному реакционному центру. Легко образуют ковалентные связи. Наиболее известными электрофилами в биохимических реакциях являются Н+, ионы металлов, углерод карбонильной группы.
Группы радикалов аминокислот – плохие электрофилы.
По числу молекулреакции можно разделить на мономолекулярные и бимолекулярные. В биологических системах реакции с большим числом участвующих молекул практически не встречаются.
По направлению реакций с учетом конечного результата можно выделить следующие типы реакций
1. Окислительно-восстановительные реакции включают потерю или присоединение электронов, в результате чего меняется степень окисления атомов, которые являются реакционными центрами. Многие окислительно- восстановительные реакции в клетке включают разрыв С-Н связи с отнятием у атома углерода двух электронов и переносе их на акцептор, роль которого могут выполнять коферменты. Конечный акцептор электронов у аэробных организмов кислород, представляющий бирадикал с двумя неспаренными электронами, Молекула кислорода поэтому может принимать только неспаренные электроны. Отнимаемые у субстрата пары электронов должны быть перенесены на кислород при помощи специальной электронпереносящей системы, включающей ряд переносчиков.
2. Реакции кислотно-основного взаимодействия. Взаимодействие между кислотами и основаниями с образованием солей
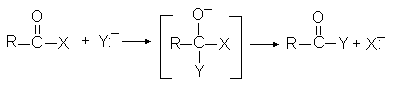
3. Реакции замещения. В ходе реакции происходит замена атомов или групп атомов на другие атомы или группы. Особое место среди реакций этой группы среди биохимических реакций занимают реакции переноса групп. Они включают перенос электрофильной группы от одного нуклеофила на другой. Такие реакции можно назвать реакциями нуклеофильного замещения. Наиболее часто переносимыми группами в биохимических реакциях являются ацильная, фосфатная и гликозильные группы. Примером такой реакции может быть гидролиз пептидной связи при помощи химотрипсина.
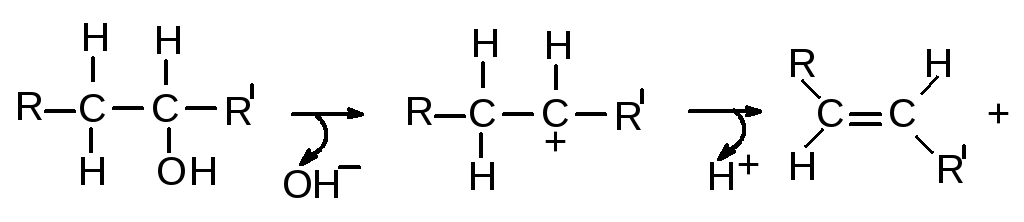
4. Реакции отщепления. Протекают одновременно по двум соседним атомам с отщеплением групп и образованием двойной связи. Отщепляемыми группами могут быть небольшие молекулы типа Н2О,NН3. Распад связи может проходить по одному из трех механизмов: а)согласованный механизм, б)постепенно с разрывом С-О связи и образованием карбокатиона, в) постепенно, с разрывом С-Н связи и образованием карбаниона. Ферменты катализируют отщепление воды двумя механизмами или протонированием ОН группы кислотой (кислотный катализ) или отщепление протона (основной катализ). Постепенное отщепление требует стабилизации образующихся заряженных групп противоположными по заряду группами (электростатический катализ). Одна из самых интересных реакций дихотомического процесса, катализируемая енолазой относится к этому типу реакций.
5. Реакции перегруппировки
изменяют форму углеродного скелета молекул. Среди метаболических реакций такой тип реакций встречается довольно часто. Например, реакции изомеризации. В результате реакции происходит перераспределение электронной плотности и характера связей.
Могут включать внутримолекулярный сдвиг водородного атома, таким образом, чтобы изменить расположение двойной связи. В таких реакциях протон отнимается у одного углеродного атома и переносится к другому. Наиболее распространенная реакция изомеризации - взаимное превращение альдоз в кетозы
Рацемизация - реакция изомеризации, в которой водородный атом изменяет свое стереохимическое положение в молекуле в хиральном центре. Эпимеризация то же самое, но молекула имеет несколько хиральных центров
6. Реакции, которые образуют или разрывают С-С связи. Это реакции, лежащие в основе и катаболизма и анаболизма. При превращении глюкозы в СО2протекает 5 реакций разрыва С-С связей. Синтез глюкозы требует столько же реакций образования этих связей. Наиболее часто в этих реакциях принимает участие электрофильный карбонильный углеродный атом. Примерами могут быть реакции альдольной конденсации, катализируемые альдолазой, декарбоксилирование-кетокислот (изоцитратдегидрогеназная реакция) и др.
Факторы, определяющие активность ферментов [E], [S], [P], Km. Влияние pH, [P], tº, ионной силы на активность ферментов.
У каждого фермента существует свой pH-оптимум. Существенное влияние на активность ферментов оказывает реакция среды. Для проявления их оптимального действия чаще всего существует узкий диапазон измерения pH среды (pH-оптимум). В некоторых случаях сдвиг pH на единицу снижает активность на 80%. Поэтому в экспериментальных условиях работы с ферментом очень важно поддерживать pH на постоянном уровне.
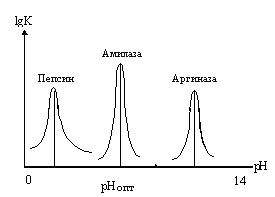 Слева приведены
кривые зависимости активности фермента
от изменения pH среды. Пепсин желудочного
сока, который катализирует гидролиз
белков, имеет pH-оптимум при pH=2. При pH=3
он теряет половину активности, а при
pH=4 активность пепсина не измеряется.
pH-оптимум амилазы, фермента, катализирующего
расщепление крахмала, составляет 7,0. И
в этом случае изменения pH на единицу
приводит к снижению активности на 50%,
а - на 2 единицы отмечается полная потеря
активности. Относительно редко
встречаются случаи, когда pH-оптимум
активности ферментов находится в сильно
кислой или щелочной среде. Это характерно
для таких ферментов как пепсин и аргиназа
(катализирует расщепление аргинина).
Большинство белков при экстремальных
значениях pH неустойчивы. рH-оптимум
является важной характеристикой
фермента, но ни как не исключительной,
присущей только данному ферменту.
Слева приведены
кривые зависимости активности фермента
от изменения pH среды. Пепсин желудочного
сока, который катализирует гидролиз
белков, имеет pH-оптимум при pH=2. При pH=3
он теряет половину активности, а при
pH=4 активность пепсина не измеряется.
pH-оптимум амилазы, фермента, катализирующего
расщепление крахмала, составляет 7,0. И
в этом случае изменения pH на единицу
приводит к снижению активности на 50%,
а - на 2 единицы отмечается полная потеря
активности. Относительно редко
встречаются случаи, когда pH-оптимум
активности ферментов находится в сильно
кислой или щелочной среде. Это характерно
для таких ферментов как пепсин и аргиназа
(катализирует расщепление аргинина).
Большинство белков при экстремальных
значениях pH неустойчивы. рH-оптимум
является важной характеристикой
фермента, но ни как не исключительной,
присущей только данному ферменту.
|
Табл 2-2.Значения pHопт. некоторых ферментов | |
|
Фермент |
pH опт. |
|
Липаза (подж.железа) |
8.0 |
|
Липаза (желудок) |
4.0 - 5.0 |
|
Липаза(касторовое масло) |
4.7 |
|
Пепсин |
1.5 - 1.6 |
|
Трипсин |
7.8 - 8.7 |
|
Уреаза |
7.0 |
|
Инвертаза |
4.5 |
|
Мальтаза |
6.1 - 6.8 |
|
Амилаза (подж.железа) |
6.7 - 7.0 |
|
Амилаза (солод) |
4.6 - 5.2 |
|
Каталаза |
7.0 |
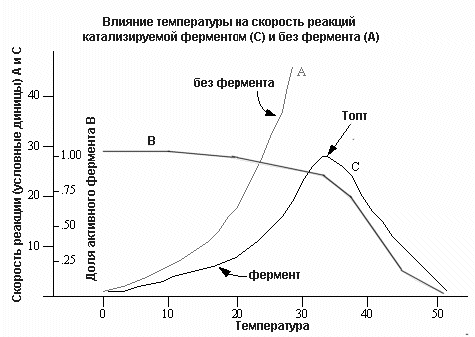
Влияние температуры на активность фермента
Дело в том, что многие ферменты имеют схожие значения pH-оптимумов. Механизм влияния pH среды на активность ферментов заключается в том, что изменение этого показателя приводит к изменению степени ионизации амино- (NH3) и карбосильных (СОО-) групп в молекуле фермента. В результате белковая молекула фермента подвергается конформационным перестройкам, что сказывается на взаимоотношения между ферментом и субстратом.
Повышение температуры неоднозначно влияет на активность фермента
Ферментные реакции, как и любое химическое превращение, в значительной степени зависят от температурных условий. Старое правило о том, что повышение температуры на 100 С удваивает скорость химической реакции распространяется и на активность ферментов.
Так как все ферменты являются белками, а белки при температуре выше 40-500С в большинстве своем необратимо изменяются, температурный интервал для работы ферментов ограничивается определенными пределами.. Активность фермента повышается при повышении температуры. Начиная с определенной температуры, совпадающей с началом денатурации белка, активность фермента падает. Следует учитывать, что стабильность отдельных ферментов при нагревании различна. Это обстоятельство находит практическое применение при выделении и очистке ферментов. В смеси ферментов с различной температурной чувствительностью можно измерить активность более устойчивого к нагреванию фермента, если предварительно нагреть смесь до температуры, разрушающей другие ферменты этой смеси. Используя это свойство, например, в клинической практике удается разделить разные изоферменты ЛДГ. Так специфический для сердца изофермент ЛДГ значительно менее чувствителен к нагреванию, чем другие изоферменты. Поэтому после нагревания реакционной смеси, остаточная активность ЛДГ отражает активность ЛДГ, в то время как ферменты, специфичные для мышц и печени при этом разрушаются, так как активность ферментов чувствительна к температуре, ферментативные реакции следует проводить при определенной температуре, обычно работают при температурах между 250и 400С.
Ферменты характеризуются высокой специфичностью
Специфичность - одно из удивительных свойств ферментов, обусловленное их белковой природой. Уже первые исследователи механизма действия ферментов отмечали это уникальное свойство ферментов, отличающее их от известных в то время катализаторов. Не вызывало сомнений и то что фермент действует не всей своей поверхностью, а свои функции выполняет при помощи отдельных участков этой поверхности. Участок молекулы фермента, обеспечивающий выполнение основных функций фермента получил название активного центра. Э. Фишер, пораженный этим свойством ферментов, предложил для объяснения специфичности взаимодействия фермента и субстрата модель «ключа и замка».
Эта модель предполагала существование такого центра как особой структуры и без связи с субстратом. Жесткие рамки взаимоотношений ключа и замка были дополнены моделью индуцированного взаимодействия, предложенной Д. Кошландом, моделью «рука-перчатка». Эта модель была подтверждена при помощи метода рентгеноструктурного анализа, позволившего построить пространственную модель фермента. Первым ферментом, пространственная структура которого была исследована этим методом, был лизоцим. Этот фермент катализировал расщепление полисахаридов стенки бактерий и некоторых грибов. Он был выделен из яиц, слюны и других биологических жидкостей. Лизоцим состоит из 129 аминокислот. В 1965 году Д.Филлипс расшифровал пространственную структуру лизоцима. Молекула представляла элипсоид вращения с размерами 30х30х45 ангстрем. В лизоциме были выявлены 5 -спиральных и три-структурных сегмента. Исследователи сразу обратили внимание на проходящую поперек молекулы глубокую расщелину, в которой помещались 6 моносахаридов субстрата. При этом 4 из 6 моносахаридов были комлементарны форме активного центра и хорошо размещались в активном центре, а для размещения двух моносахаридов потребовалось изменить их конформацию. В активном центре лизоцима были выявлены аминокислотные остатки, обеспечивающие связывание фермента и субстрата и аминокислотные остатки, обеспечивающие проведение реакции. В последующем такие исследования были проведены со многими ферментами, что позволило выявить в активном центре целый ряд функциональных групп или участков:
Способствующие группы– обеспечивают пространственную ориентацию субстратов уже при сближении фермента и субстрата, что позволяет субстрату взаимодействовать с активным центром фермента в наиболее благоприятной конформации.
Якорный или субстрат связывающий участок– обеспечивает наиболее адекватное и прочное связывание субстрата в активном центре фермента.
Химические связи, стабилизирующие пространственную структуру белков, природа применила и для взаимодействия фермента и его субстрата. К этим связям следует отнести прежде всего нековалентные связи и взаимодействия. Это
1. Силы Ван дер Ваальса
2. Электростатическое взаимодействие
3. Водородные связи
4. Гидрофобные взаимодействия
Каталитический участок- формируется группами , обеспечивающими проведение катализа
Вспомогательные группы– группы, повышающие реакционоспособность каталитического участка.
|
|
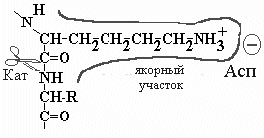
Якорный участок трипсина представлен длинным узким карманом с отрицательно заряженный Асп в глубине кармана. В этой карман легко проникают аминокислоты, имеющие длинную боковую цепь с положительным зарядом на конце; такими аминокислотами являются Лиз или Ар, которые хорошо связываются и распознаются, а гидролиз происходит на соседней пептидной связи. |
|
|
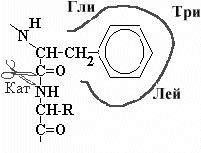
В гидрофобном кармане, образованном радикалами Гли, Три и Лей химотрипсина располагается боковая цепь с ароматическим кольцом (Фен, Тир или Три).. Пептидная связь образованная СООН группой ароматической аминокислоты устанавливается рядом с каталитическим участком химотрипсина. |
|
|
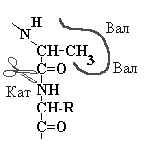
Фермент эластаза подобен химотрипсину, но в эластазе, часть кармана блокирована парой объемистых валинов. Наиболее комплементарным этому карману может быть участок пептида содержащий Ала или Гли |
Пространственная структура групп формирующих активный центр и является структурной основой специфичности. Специфичность у разных ферментов может проявляться по-разному.Ферменты как белки, построены изL-аминокислот и эта особенность придает ферментамстереохимическую специфичность. Такие ферменты взаимодействуют и катализируют превращения только одного из стерических или оптических изомеров субстрата. Например, одни оксидазы аминокислот избирательно действуют наL-аминокислоты, а другие только наD-аминокислоты. Помимо стереохимической специфичности многие ферменты обладают высокой избирательностью к типу химических групп субстратов. В большинстве случаев избирательность к группам – геометрическая специфичность – даже выше чем стереохимическая специфичность. Правда, лишь небольшая часть ферментов обладаетабсолютной специфичностью, т.е. катализирует превращение только одного субстрата. Чаще всего ферменты обладаютгрупповой специфичностью.Это означает, что они действуют на группу субстратов, предъявляя требования к типу группы и типу связи–абсолютная групповаяспецифичность или только к типу связи –относительная групповаяспецифичность. Нередко в объяснении типа специфичности используют термин предпочтение. Например, карбоксипептидаза А катализирует гидролиз всех С-концевых пептидных связей за исключением связей образованных Лиз, Арг или Про, если предшествующая аминокислота не Про. Обладая выраженной специфичностью к одному и тому же субстрату, ферменты могут отличаться по типу реакций, которые они катализируют. Например, глюкоза-6-фосфат – субстрат многих ферментов и глюкозо-6-фосфтазы и дегидрогеназы глюкозо-6-фосфата и др. Подобные взаимоотношения складываются у ферментов и с коферментами. Например, существуют дегидрогеназы, которые катализируют реакции окисления только при соединении с коферментом – нуклеотидом НАД+. С НАДФ+, который отличается только одной дополнительной фосфатной группой такие дегидрогеназы не взаимодействуют. И наоборот, есть дегидрогеназы, которые взаимодействуют только с НАДФ+. Некоторые ферменты не проявляют специфичности к типу связи, что нашло применение в лабораторной практике. Многие протеолитические ферменты катализируют гидролиз не только пептидных связей, но и эфирные связи. И если методически количественно трудно оценить скорость реакции гидролиза пептидных связей, то использование синтетических субстратов, представляющих эфиры аминокислот с красящимися веществами, позволяют быстро и просто оценить активность таких ферментов в клинической биохимической лаборатории.
Изостерическая и аллостерическая регуляция.
Регуляция активности ферментов бывает пассивная (с помощью изменения условий среды) т. е. есть постоянные ферменты и непостоянные, которые появляются под действием каких-либо факторов среды. (Под действием температуры или с помощью ионной силы и pH, [S], [E]).
Активная регуляция:
|
изостерическая; |
аллостерическая. |
|
(регуляция с помощью субстрата и продукта) |
(регуляция активности фермента с помощью веществ, отличных от S и P). |
Регуляция путем изменения количества фермента.
У бактерий хорошо изучен феномен индуцированного синтеза ферментов при выращивании на средах с одним углеводом, например, глюкозой. Замена глюкозы на лактозу приводит к индуцированному синтезу фермента галактозидазы, расщепляющей лактозу на глюкозу и галактозу.
В животных тканях подобный быстрый синтез ферментов наблюдается реже, однако при поступлении в организм некоторых ядов, канцерогенных веществ, алкалоидов наблюдается резкое увеличение количества (а значит и активности) гидроксилаз, окисляющих чужеродные вещества в нетоксичные продукты. С другой стороны, иногда под действием этих гидроксилаз чужеродные вещества превращаются в более токсичные продукты (летальный синтез).
Регуляция активности по принципу обратной связи.
Допустим в клетке есть многоступенчатый биосинтетический процесс, каждая стадия которого катализируется собственным ферментом:
E1 E2 E3 E4
A ---> X ---> Б ---> B ---> Г ---> ... P
Накопление продукта P оказывает мощное ингибирующее действие на фермент E1.
Аллостерическая регуляция.
Аллостерические ферменты - это ферменты, располагающиеся в начале метаболического потока или на его узловых этапах и управляют этим метаболическим потоком.
Свойства аллостерических ферментов:
1. Являются олигомерами состоящими из протомеров.
2. Имеют как минимум два центра: активный центр и центр аллостерической регуляции.
3. Имеют ось симметрии.
4. Протомеры изменяют свою структуру в пределах олигомеров.
5. Изменение конформации олигомеров ограничено конформациями отдельных протомеров.
Существует 2 вида веществ (эффекторы), которые оказывают на фермент двоякое действие:
1) активаторы; 2) ингибиторы.
Аллостерический фермент имеет 2 центра аллостерической регуляции - центр аллостерической активации
- центр аллостерического ингибирования.
При взаимодействии аллостерического фермента с аллостерическим активатором резко возрастает степень сродства фермента к субстрату, точнее возрастает степень сродства активного центра к субстрату.
При взаимодействии аллостерического ингибитора с аллостерическим ферментом, резко понижается степень сродства фермента к субстрату.
Наличие двух центров в аллостерическом ферменте доказывается путем технической денатурации в мягких условиях (под действием мочевины) ---> при этом аллостерический фермент теряет регуляторные свойства (они связаны с центрами аллостерической регуляции), но сохраняет каталитические свойства, связанные с активным центром.
|
Заведующий кафедрой биологической химии, д.м.н., проф. |
Грицук А. И. |
___________ |
21.10.2006
Министерство здравоохранения Республики Беларусь
УО «Гомельский государственный медицинский университет»
Кафедра биологической химии
Обсуждено на заседании кафедры (МК или ЦУНМС)
Протокол № _________________200__года
ЛЕКЦИЯ по биологической химии
наименование дисциплины
для студентов _2__ курсалечебногофакультета
Тема Ферменты 3. Регуляция активности.
Время 90 мин.
Учебные и воспитательные цели:
Дать представление:
О механизмах и роли аллостерической регуляции. Характеристике аллостерических ферментов. Видах ингибирования (обратимое, необратимое, конкурентное, неконкурентное, бесконкурентное).
О регуляции активности ферментов путем химической модификации: Реакциях ограниченного протеолиза, аденилирования, рибозилирования, ацетилирования, фосфорилирования (роли гормонов, АЦ-комплекса, цАМФ, цГМФ, ионов Ca).
Об изоферментах, их природе, биологической роли, строении ЛДГ.
Об изменении активности ферментов в онтогенезе.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф.Биологическая химия. М.: Медицина, 1990. С. 126–132; 1998. С. 157–168.
Николаев А. Я.Биологическая химия. М.: Высшая школа, 1989. С. 52–92.
Дополнительная
Марри Р. и др. Биохимия человека. М.: Мир, 1993. Т. 1. С. 63–75.
Филиппович Ю. Б. Основы биохимии. М.: Высшая школа, 1993. С. 105–144.
Врожденные и приобретенные энзимопатии / Под ред. Ташева Т. М.: Медицина, 1980.
Вилкинсон Д. Принципы и методы диагностической энзимологии. М.: Медицина, 1981.
Руководство по клинической лабораторной диагностике. Киев: Вища школа, 1990. С. 167–186.
Зилва Ф., Пеннел Дж. Клиническая химия в диагностике и лечении. М.: Медицина, 1986. С. 372–388.
МАТЕРИАЛЬНОЕ ОБЕСПЕЧЕНИЕ
1. Мультимедийная презентация.
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№ п/п |
Перечень учебных вопросов |
Количество выделяемого времени в минутах |
|
|
Механизмы и роль аллостерической регуляции. Характеристика аллостерических ферментов. Виды ингибирования (обратимое, необратимое, конкурентное, неконкурентное, бесконкурентное). |
30 |
|
|
Регуляция активности ферментов путем химической модификации: Реакции ограниченного протеолиза, аденилирования, рибозилирования, ацетилирования, фосфорилирования (роль гормонов, АЦ-комплекса, цАМФ, цГМФ, ионов Ca). |
30 |
|
|
Изоферменты, их природа, биологическая роль, строение ЛДГ. |
20 |
|
|
Изменение активности ферментов в онтогенезе. |
10 |
Всего 90 мин
Механизмы и роль аллостерической регуляции. Характеристика аллостерических ферментов. Виды ингибирования (обратимое, необратимое, конкурентное, неконкурентное, бесконкурентное).
Кинетика многих ферментов не подчиняется принципам кинетики Михаэлиса и Ментен
При знакомстве с материалом по кинетике ферментов могло сложиться впечатление, что кинетика всех ферментов основана на принципах Михаэлиса и Ментен. Исключением было только влияние избытка субстрата. У ферментов, подчиняющимся принципам кинетики Михаэлиса и Ментен с позиций процессов, протекающих в клетке, имеется ряд недостатков. Для обеспечения нормальной жизнеспособности клетки требуется тонкая регуляция концентрации большинства метаболитов. Существует довольно узкий диапазон допустимых концентраций субстратов в клетке при потребности быстро реагировать изменениями скоростей реакции в широких пределах в ответ на постоянно меняющееся функциональное состояние клетки. Приведенный ранее расчет показывает, что скорости реакций мало чувствительны к изменениям концентрации субстрата. Например, повышение скорости от 0,1Vmaxдо 0.9Vmaxтребует повышения концентрации субстрата в 81 раз. Такие же цифры характеризуют и действие ингибиторов. Ферменты, управляющие превращениями метаболитов должны быть более чувствительны к изменению концентраций и субстрата и ингибитора. Таким образом, природа вынуждена обратиться к "кооперативным" системам, в которых маленькие изменения в одном параметре, например концентрации ингибитора, вызывают большие изменения в скорости. Графически результат работы такой кооперативной системы (график зависимости скорости реакции от концентрации субстрата) выражается не гиперболой, аS-образной сигмоидной кривой (сигмоидальные кривые всегда сдвинуты вправо в сравнении с гиперболой). Существует большое семейство ферментов, которые представляют такие кооперативные системы. Это аллостерические ферменты.
Определение.Аллостерический белок определяется как белок, содержащий два или больше топологически различающихся центра связывания лигандов (субстраты, ингибиторы и т.д), которые функционально взаимодействуют друг с другом. Связывание лиганда, с одним центром изменяет свойства другого (их).
Большая часть аллостерических белков – аллостерические ферменты, но некоторые белки, типа гемоглобина, выполняют и другие функции.
Кооперативность Кооперативность - модификация константы связывания лиганда белком предшествующим связыванием другого лиганда. Константы связывания - подобны Ks для субстрата или Ki для ингибитора, являются в основном константами диссоциации белка и лиганда и указывает силу связывания, или сродство белка и лиганда. Км обычно принимается как константа связывания субстрата, поскольку ее проще измерять, чем Ks. Понятие кооперативность означает, что связывание одного лиганда к белку или увеличит или уменьшит способность белка, связывать вторую молекулу лиганда. Если модификация увеличивает способность связывания (или сродство), это называют положительной кооперативностью. Если способность связывания снижается - это отрицательная кооперативность. Два лиганда один из которых влияет на связывание другого, могут быть химически идентичны, например, одна молекула субстрата, изменяет связывание другой молекулы субстрата. Такое взаимодействие называют гомотропным эффектом. Если они химически различаются, например, влияние ингибитора на связывание субстрата, тогда это - гетеротропный эффект.
У аллостерических ферментов особые свойства
Аллостерические ферменты обладают рядом свойств, которые отличают их от не аллостерических. Следует подчеркнуть, что приведенные ниже свойства не обязательны для всех аллостерических белков. Это – общие особенности, по крайней мере некоторые из них проявляются у отдельных аллостерических белков.
Полимерная структура
Сигмоидная ( в отличие от гиперболической для не аллостерических) форма кривой зависимости скорости реакции от концентрация субстрата
Существование эффекторов
Двухфазный ответ на конкурентные ингибиторы
Потеря аллостерических свойств при денатурации
Полимерная структура. Все аллостерические ферменты и белки имеют полимерную или четверичную структуру. Это значит, что типичный аллостерический белок будет состоять из ряда отдельных белковых цепей или субъединиц, которые связаны друг с другом слабыми взаимодействиями типа водородных связей и гидрофобного взаимодействия. Полимерная структура – ключевое свойство для функции аллостерического белка.
|
|
|
|
Кооперативное связывание |
Не кооперативное связывание |
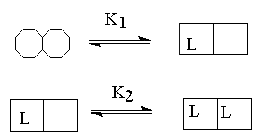
Аллостерический фермент содержит ряд активных центров, в самом простом случае по одному на субъединицу, каждый из которых может связываться с лигандом. Взаимодействие между этими центрами и является основой кооперативности. Так, в типичном аллостерическом ферменте связывание молекулы лиганда к одному из активных центров инициирует изменение конформации, которое увеличит способность других активных центров связывать лиганды (положительная кооперативность. K2 <<K1) или понизит их сродство к лиганду (отрицательная кооперативностьK1<<K2). При отсутствии кооперативного взаимодействия -K1=K2.
Субъединицы
связаны друг с другом слабым
взаимодействием, поэтому аллостерический
фермент будет часто существовать в
растворе в равновесии между целым
ферментом и индивидуальными субъединицами.
При большом числе субъединиц могут
возникать промежуточные формы между
этими крайними формами, в которых будут
связаны несколько субъединиц, но в
меньшем количестве, чем в целом
ферменте. В этом случае одиночная
субъединица не может быть каталитически
активна. Самая маленькая каталитически
активная структура названа протомером.
Связывание лигандов (субстрат, продукт
или эффектор) к ферменту изменяет
позицию равновесия между субъединицами.
Сигмоидная кинетика.При
составлении графика зависимости
скорости от концентрации субстрата
для ферментативной реакции для
аллостерических ферментов получается
несколько иной тип кривой, называемой
сигм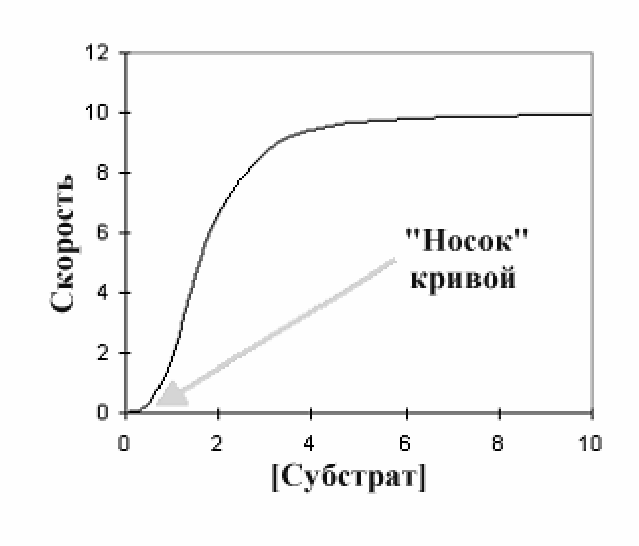 оидальной
в отличие от гиперболической для не
аллостерических ферментов. Ключевой
элемент этой кривой, который отличает
ее от гиперболической - «носок» у
основания кривой , который можно видеть
при низких концентрациях субстрата.
В этой точке увеличение концентрации
субстрата вызывает очень незначительное
увеличение в скорости - диаграмма имеет
очень небольшой наклон. При более
высоких уровнях субстрата, (выше 0.5
единиц концентрации на этом графике)
можно видеть, что увеличение концентрации
субстрата начинает вызывать намного
более значительное увеличение в
скорости, и кривая становится более
крутой. При высоких концентрациях
субстрата график становится очень
похожим на гиперболический график, и
можно видеть такую же пологую кривую
как и на графике уравнения Михаэлиса
и Ментен.
оидальной
в отличие от гиперболической для не
аллостерических ферментов. Ключевой
элемент этой кривой, который отличает
ее от гиперболической - «носок» у
основания кривой , который можно видеть
при низких концентрациях субстрата.
В этой точке увеличение концентрации
субстрата вызывает очень незначительное
увеличение в скорости - диаграмма имеет
очень небольшой наклон. При более
высоких уровнях субстрата, (выше 0.5
единиц концентрации на этом графике)
можно видеть, что увеличение концентрации
субстрата начинает вызывать намного
более значительное увеличение в
скорости, и кривая становится более
крутой. При высоких концентрациях
субстрата график становится очень
похожим на гиперболический график, и
можно видеть такую же пологую кривую
как и на графике уравнения Михаэлиса
и Ментен.
Такой тип диаграммы характерен для положительной субстратной кооперативности. При очень низких концентрациях субстрата лишь небольшое количество активных центров фермента связываются с субстратом и фермент будет иметь низкое сродство к субстрату. Поэтому добавление большего количества субстрата вызывает только небольшое увеличение в скорости реакции, поскольку субстраты связываются очень плохо. Однако, по мере повышения числа связавшихся молекул субстрата, положительный кооперативный эффект увеличивает способность фермента связывать субстраты, и кривая на графике начинает это показывать, круто перемещаясь вверх. В конечном счете, точно так же как и в случае классической гиперболической кривой, ферменты постепенно насыщаются субстратом и линия делается пологой, показывая достижение максимальной скорости. Такой тип кривой не обязателен для всех аллостерических ферментов. В частности, многие ферменты катализируют превращение нескольких субстратов и исследования показывают, что такие ферменты могут проявлять положительную кооперативность (сигмоидальная кривая) для одного субстрата и катализировать реакцию по гиперболической кривой для другого.
Эффектор. Аллостерические ингибиторы и активаторы объединяют общим названием эффекторы. Эффектор - одна из важных особенностей аллостерических ферментов. Возможность изменять скорость реакции, катализируемой ферментом, ингибиторами и активаторами - краеугольный камень принципов регуляции метаболизма. Следующий график показывает пути, по которым эффекторы изменяют кинетический график типичного аллостерического фермента с положительной субстратной кооперативностью.
Ц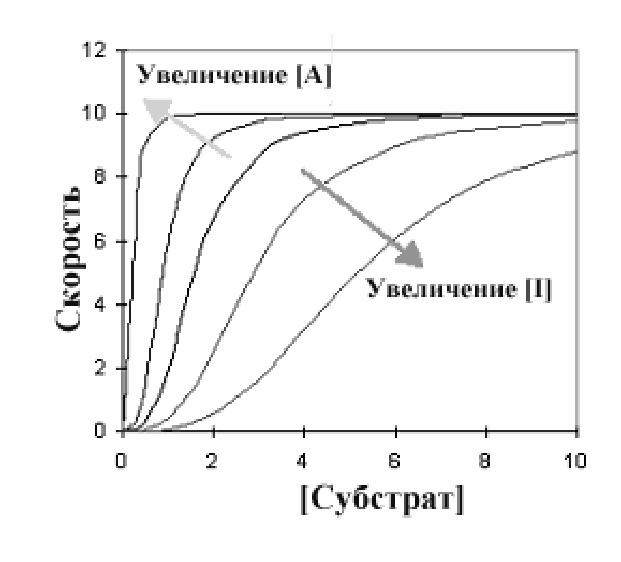 ентральная
линия графика - типичная сигмовидная
кривая в отсутствии любого эффектора.
В присутствии активатора (А) скорость
реакции повышается при любой данной
концентрации субстрата, в то время как
ингибитор уменьшает скорость реакции.
Интересны и изменения общей формы
кривой по сравнению с центральной
линией. Ингибитор увеличил сигмовидную
форму, удлиняя «носок» кривой, в то
время как активатор оказывал
противоположный эффект. При более
высокой концентрации активатора график
в целом приобретает характер гиперболы.
Это указывает на то, что аллостерический
ингибитор увеличивает уровень субстратной
кооперативности, в то время как активатор
уменьшает его.
ентральная
линия графика - типичная сигмовидная
кривая в отсутствии любого эффектора.
В присутствии активатора (А) скорость
реакции повышается при любой данной
концентрации субстрата, в то время как
ингибитор уменьшает скорость реакции.
Интересны и изменения общей формы
кривой по сравнению с центральной
линией. Ингибитор увеличил сигмовидную
форму, удлиняя «носок» кривой, в то
время как активатор оказывал
противоположный эффект. При более
высокой концентрации активатора график
в целом приобретает характер гиперболы.
Это указывает на то, что аллостерический
ингибитор увеличивает уровень субстратной
кооперативности, в то время как активатор
уменьшает его.
K-системы и V-системы Как видно из графика все линии стремятся к одному значениюVmax, но при этом эффектор влияет на связывание субстрата, что отмечено в изменении Км. Такая ситуация названаK-системой. Некоторые ферменты имеют эффекторы, которые изменяютVmax. Тогда говорят оV-системе..
Двухфазный ответ на конкурентные ингибиторы.Помимо взаимодействия с эффекторами, аллостерические ферменты являются объектом обычного конкурентного торможения, подобно любому другому ферменту. Классические конкурентные ингибиторы действуют, потому что они структурно подобны субстрату фермента. В аллостерическом ферменте, с положительной субстратной кооперативностью конкурентный ингибитор также достаточно близок по строению субстрату, мог бы иметь те же самые свойства кооперативности как и субстрат. В этом случае, низкая концентрация конкурентного ингибитора увеличивает способность фермента связывать молекулы субстрата, что фактически равно увеличению скорости реакции. При более высоких концентрациях ингибитора это блокировало бы связывание субстрата обычным способом, и реакция замедлится. Ингибитор тем самым оказывает двухфазный эффект. При низких концентрациях - он действует как активатор, в то время как при высоких концентрациях, он действует как ингибитор.
Денатурирующие агенты. Денатурация – нарушение пространственной структуры фермента с последующей потерей активности фермента. Денатурация вызывается рядом факторов, включая высокую температуру, экстремальные значения рН и химические денатурирующие реактивы типа мочевины.
Аллостерические ферменты, подвергнутые умеренному воздействию одним из этих факторов денатурации часто вначале теряют свои аллостеричесие свойства (субстратную кооперативность) при сохранении способности катализировать реакции. Это хорошее доказательство того, что третичная структура играет ведущую роль не только для механизмов катализа, но для механизмов аллостерической регуляции.
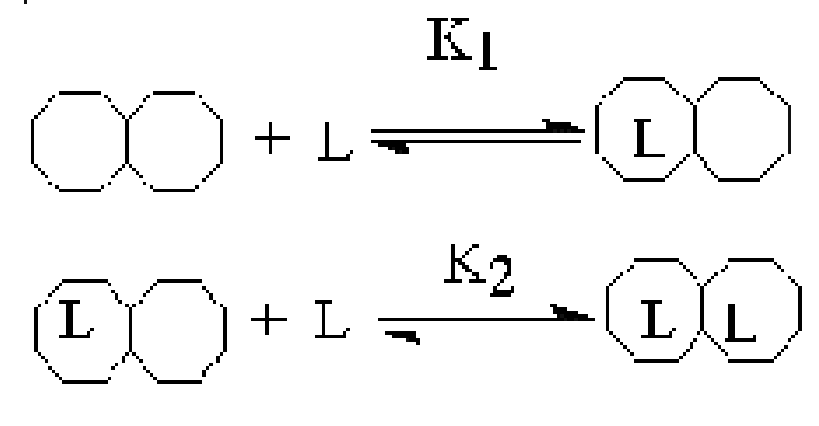
Две модели объясняют механизмы аллостерии.
Предложены две основные модели, описывающие механизмы аллостерии: согласованный механизм аллостерических взаимодействий или симметричная модель, предложенная Жаком Моно, Джефри Уайменом и Жаном-Пьером Шанже в 1965 году , и последовательная модель, предложенная Даниелом Кошландом. В основном эти модели лишь крайние формы представлений о механизме работы аллостеричеких ферментов.. Главное различие между ними в том, что симметричная модель представляет мультимерный аллостерический фермент как объединение нескольких субъединиц, согласованно изменяющих свою конформацию, в то время как модель Кошланда учитывает сосуществование смешанных, или гибридных молекул, субъединицы которых могут находится в различных конформационных состояниях.
Симметричная модель. Симметричная модель аллостерии является простым и изящным объяснением положительной субстратной кооперативности и влияния аллостерических эффекторов. Она основана на идее, что аллостерический фермент состоит из ряда субъединиц, которые могут существовать в двух различных конформациях. Их обозначают как расслабленное илиR-состояние, и напряженное илиT-состояние.
В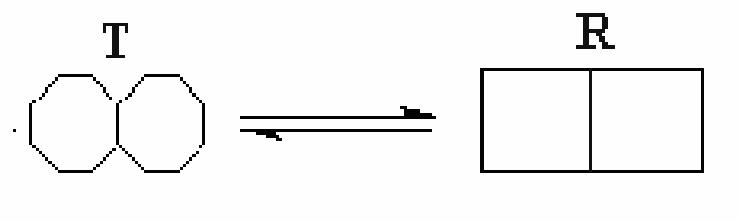 се
субъединицы данной молекулы фермента
должны иметь одну и ту же конформацию.
Другими словами, молекула фермента
должна состоять полностью изRсубъединиц илиTсубъединиц,
и не может содержать смесь этих форм.
В растворе эти две формы находятся в
равновесии. Поскольку смесей типов
субъединиц в индивидуальном белке
невозможны, в растворе возникает
простое равновесие между белками,
состоящими полностью изRиTсубъединиц.
се
субъединицы данной молекулы фермента
должны иметь одну и ту же конформацию.
Другими словами, молекула фермента
должна состоять полностью изRсубъединиц илиTсубъединиц,
и не может содержать смесь этих форм.
В растворе эти две формы находятся в
равновесии. Поскольку смесей типов
субъединиц в индивидуальном белке
невозможны, в растворе возникает
простое равновесие между белками,
состоящими полностью изRиTсубъединиц.
Для объяснения работы модели нужно сделать еще два предположения:
Во-первых в растворе и в отсутствии любых лигандов (субстрата или аллостерического эффектора) равновесие сдвинуто в сторону T-формы.
Во-вторых, конформация активных центров такова, что R-состояние имеет более высокое сродство к лиганду. Это не обязательно означает, чтоTформа неспособна к связыванию лиганда, просто у нее более низкое сродство.
Е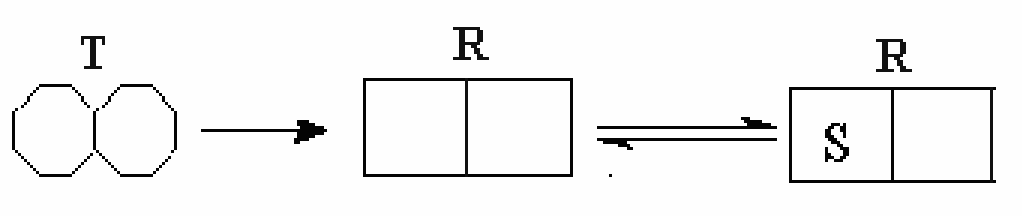 сли
добавляется небольшое количество
лиганда к этому раствору, более вероятно,
что он свяжется сRбелком,
поскольку тот имеет более высокое
сродство. Это приведет к образованию
комплекса, в котором белокRсо связанной молекулой лиганда, утратит
способность переходить в Т форму, что
сместит сложившееся равновесие вправо,
увеличив числоRбелков
с высоким сродством к лиганду и увеличив
тем самым общее сродство к лиганду в
системе. Результат - положительная
кооперативность - связывание одной
молекулы лиганда а увеличивает
способность белка, связывать другие
молекулы лигандов.
сли
добавляется небольшое количество
лиганда к этому раствору, более вероятно,
что он свяжется сRбелком,
поскольку тот имеет более высокое
сродство. Это приведет к образованию
комплекса, в котором белокRсо связанной молекулой лиганда, утратит
способность переходить в Т форму, что
сместит сложившееся равновесие вправо,
увеличив числоRбелков
с высоким сродством к лиганду и увеличив
тем самым общее сродство к лиганду в
системе. Результат - положительная
кооперативность - связывание одной
молекулы лиганда а увеличивает
способность белка, связывать другие
молекулы лигандов.
А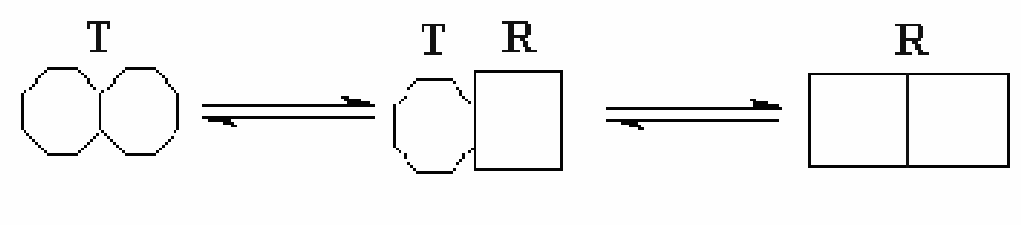 ллостерические
эффекторы.Влияние эффекторов на
ферменты можно объяснить просто,
добавив к сказанному выше, что у фермента
есть два центра связывания лигандов.
Роль одного лиганда будет выполнять
субстрат, взаимодействующий с активным
центром, а второй лиганд – эффектор,
связывающийся со специальным
аллостерическим центром. Субъединицы
фермента также могут принимать илиRилиTконформации. Активатор
хорошо связывается с ферментом, который
находится вR-конформации,
обладающей одновременно высоким
сродством к субстрату. Связывание
активатора смещает равновесие в сторонуRконформации с высоким
сродством к субстрату, что способствует
повышению скорости реакции.(«носок»
на сигмовидной кривой). Чем выше
концентрация активатора, тем выше
скорость реакции и тем более похожей
на гиперболу становится график
зависимости скорости реакции от
концентрации субстрата (сглаживание
«носка» на кривой), и при больших
количествах активатора на графике
возникает гипербола. Эффектор со
свойствами ингибитора имеет более
высокое сродство кT-конформации.
Его присоединение смещает равновесие
в противоположном направлении, к
появлению в растворе ферментов с низким
сродством к субстрату и уплощению и
удлинению «носка» на графике .
ллостерические
эффекторы.Влияние эффекторов на
ферменты можно объяснить просто,
добавив к сказанному выше, что у фермента
есть два центра связывания лигандов.
Роль одного лиганда будет выполнять
субстрат, взаимодействующий с активным
центром, а второй лиганд – эффектор,
связывающийся со специальным
аллостерическим центром. Субъединицы
фермента также могут принимать илиRилиTконформации. Активатор
хорошо связывается с ферментом, который
находится вR-конформации,
обладающей одновременно высоким
сродством к субстрату. Связывание
активатора смещает равновесие в сторонуRконформации с высоким
сродством к субстрату, что способствует
повышению скорости реакции.(«носок»
на сигмовидной кривой). Чем выше
концентрация активатора, тем выше
скорость реакции и тем более похожей
на гиперболу становится график
зависимости скорости реакции от
концентрации субстрата (сглаживание
«носка» на кривой), и при больших
количествах активатора на графике
возникает гипербола. Эффектор со
свойствами ингибитора имеет более
высокое сродство кT-конформации.
Его присоединение смещает равновесие
в противоположном направлении, к
появлению в растворе ферментов с низким
сродством к субстрату и уплощению и
удлинению «носка» на графике .
Симметричная модель объясняет свойства аллостерических ферментов простым и изящным способом, однако свойства некоторых ферментов лучше объясняются последовательной моделью.
Последовательная модель. Симметричная модель была основана на условии, что одна молекула фермента содержит только один тип субъединиц -RилиT. Последовательная гипотеза обращается к возможности существования смешанных ферментов , содержащих оба типа. Равновесие лишь достигается в растворе, в котором законченныеRиTструктуры просто представляют экстремальные значения:
В основе связывания субстрата - индуцированное взаимодействие.
Последовательная модель принимает, что субстрат оказывает более прямое влияние на форму фермента. В отсутствии субстрата фермент существовал бы более или менее полностью в T-форме, которая имеет очень низкое сродство к субстрату. Так как субстрат входит в активный центр, как обычно путем случайного столкновения, отдельные части молекулы белка фермента самостоятельно обхватывают субстрат, обеспечивая хорошее взаимодействие. Это известно как индуцированное взаимодействие. Процесс индуцированного взаимодействия способствует переходу субъединицы, с которой связался субстрат вR-конформацию.
Изменение конформации одной субъединицы индуцирует изменения структуры другой
Одна субъединица теперь была преобразована в R-форму, но другие - все еще вT-состоянии. Отметим, что эти субъединицы связаны друг с другом и взаимодействуют друг с другом. Если мы принимаем, что переход одной субъединицы изTсостояния вRоказывает влияние на переход в состояниеRдругих субъединиц, мы говорим о положительной кооперативности в действии субстрата, поскольку большее количество субъединиц, вероятно, перейдет в состояние с более высоким сродством к субстрату. Это изменение для других субъединиц может происходить до связывания субстрата или просто облегчать индуцированное взаимодействие при приближении молекулы субстрата.
Ингибиторы и активаторы. Влияние аллостерических эффекторов можно также легко объяснить. Активатор подобен в своем действию субстрату только уже при связывании с другим центром на субъединице, в то время как ингибитор будет делать фермент более жестким, и затрудняет индуцированое взаимодействие при переходе от Т доR.
Отрицательная субстратная кооперативность. Отрицательная субстратная кооперативность встречается не часто, но это происходит у некоторых ферментов. Симметричная модель не может объяснить отрицательную субстратную кооперативность, так как на основе закона действующих масс трудно объяснить перемещениеRTравновесия в сторону высокого сродства. Последовательная модель объясняет это совершенно легко. Нужно только принять, что взаимодействие между субъединицами - таково, что преобразование одной из них вR-форму вызванную индуцированным взаимодействием делает это более трудным для других субъединиц.
Какая гипотеза является правильной?
Существование отрицательной кооперативности дает право предположить, что последовательная гипотеза более реальна. С другой стороны существование смесей Т и Rсубъединиц ведет к намного более сложному равновесию. Некоторые исследования, используя быстрые методы измерения скорости реакции, предполагают, что функция некоторых ферментов лучше объясняется с позиций симметричной модели. В некоторой мере гипотеза симметрии может быть рассмотрена как частный случай последовательной, в которомT/Rкомбинации могут существовать лишь на протяжении очень короткого времени.
Ингибиторы бывают разные: обратимые и необратимые
Вещества со свойствами ингибиторов ферментов можно грубо разделить на обратимые и необратимые. Обратимые ингибиторы связываются с ферментом, используя слабые связи, подобные тем, которые используются ферментом в связывании субстрата. Эти связи формируются быстро, но также быстро и легко разрушаются. Следствием такого связывания обратимого ингибитора является эффективное мгновенное действие, но после удаления ингибитора фермент сохраняет свою активность. Ингибитор находится в равновесии с ферментом, формируя комплекс ингибитора фермента:
С![]() тепень
торможения зависит от количества
фермента, связавшегося с ингибитором,
т.е. от позиции равновесия.
тепень
торможения зависит от количества
фермента, связавшегося с ингибитором,
т.е. от позиции равновесия.
Н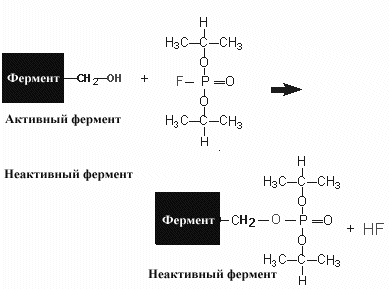
![]() еобратимые
ингибиторы известны также как инактиваторы
фермента. Они связываются с ферментом,
формируя прочные, обычно ковалентные
связи:
еобратимые
ингибиторы известны также как инактиваторы
фермента. Они связываются с ферментом,
формируя прочные, обычно ковалентные
связи:
Реакция практически необратима и фермент теряет свою активность. Учитывая, что ковалентные связи образуются медленнее, для проявления действия необратимого ингибитора требуется некоторое время для взаимодействия с ферментом. Следовательно, действие необратимого ингибитора обычно зависит от времени и степень торможения увеличивается со временем контакта его с ферментом.
Многие необратимые ингибиторы нашли применение в исследовании ферментов и медицине.
Среди примеров необратимо действующих ингибиторов можно назвать диизопропилфторфосфат (ДИПФФ). Это соединение вошло в историю энзимологии как соединение использовавшееся для исследования роли химических групп в в структуре активного центра. ДИПФФ ковалентно связывается с гидроксильной группой серина и если эта группа важна в катализе реакции, фермент терял свою активность. Эти исследования позволило выявить группу ферментов, в активном центре которых активную роль играет серин (сериновые протеазы)
Другое соединение
иодацетамид, образует ковалентную
связь с SH–группами
цистеина и если эта аминокислота важна
для активности фермента, такой фермент
утрачивает активность.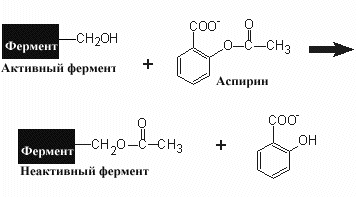
Ацетилсалициловая кислота (известный всем аспирин) является необратимым ингибитором циклооксигеназы- фермента участвующего в синтезе простагландинов. Ингибитором синтеза протеогликанов стенки бактерий является пенилиллин, структура которого напоминает D-аланин, встраиваемый в структуру протеогликанов. Связываясь с активным центром фермента бактерии благодаря своей схожести с переходным состоянием промежуточного продукта в активном центре, пенициллин образует ковалентную связь и тормозит работу фермента.
Обратимые ингибиторы могут быть конкурентными и неконкурентными
Различия между этими типами обратимых ингибиторов касаются их взаимоотношений с субстратом. Если ингибитор конкурирует с субстратом за место связывания на ферменте и действие одного может быть отменено избытком другого, говорят о конкурентном торможении, в противном случае речь идет о неконкурентных ингибиторах.
Конкурентные ингибиторы не всегда структурно подобны субстрату.
Различают два механизма конкурентного торможения.
А.Конкурентное торможение путем связывания активного центра. Классический конкурентный ингибитор - вещество, которое имеет структурное сходство с субстратом фермента. Благодаря этому подобию ингибитор может связываться с активным центром вместо субстрата. Это своеобразная молекулярная ошибка. Однако, поскольку субстрат и ингибитор не идентичны полностью, фермент не способен катализировать превращение ингибитора в продукт. Ингибитор просто блокирует активный центр фермента. Если субстрат свяжется с активным центром раньше, чем ингибитор, ингибитор не может связаться с ферментом. Нельзя одновременно обоим связаться с активным центром. Такой способ конкурентного торможения получил название изостерического из-за схожести (изос) структур субстрата и ингибитора. Наиболее часто в клетке в роли классического конкурентного ингибитора выступает продукт данной реакции, что имеет глубокий практический смысл.
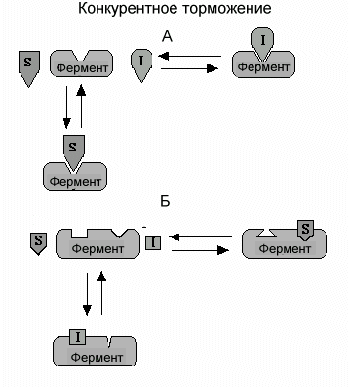 Б. Конкурентное
торможение путем изменения конформации
фермента. В отличие от классического
варианта, ингибитор связывается не с
активным центром, а со специальным
центром, связывающим ингибитор, который
расположен вдали от активного центра.
Связывание ингибитора вызывает
изменение пространственной структуры
(изменение конформации) в области
активного центра, которое не позволяет
присоединиться субстрату. Предшествующее
связывание субстрата к активному центру
в свою очередь, вызывает изменения
конформации центра связывания
ингибитора, которое предотвращает
связывание ингибитора. И субстрат и
ингибитор не могут одновременно
связаться с ферментом. В этом виде
конкурентного торможения ингибитор
может иметь любую химическую структуру,
поскольку они связываются с различными
участками фермента.
Б. Конкурентное
торможение путем изменения конформации
фермента. В отличие от классического
варианта, ингибитор связывается не с
активным центром, а со специальным
центром, связывающим ингибитор, который
расположен вдали от активного центра.
Связывание ингибитора вызывает
изменение пространственной структуры
(изменение конформации) в области
активного центра, которое не позволяет
присоединиться субстрату. Предшествующее
связывание субстрата к активному центру
в свою очередь, вызывает изменения
конформации центра связывания
ингибитора, которое предотвращает
связывание ингибитора. И субстрат и
ингибитор не могут одновременно
связаться с ферментом. В этом виде
конкурентного торможения ингибитор
может иметь любую химическую структуру,
поскольку они связываются с различными
участками фермента.
Конкурентные ингибиторы не влияют на Vmax, они понижают Км.
Реакции связывания
и субстрата и конкурентного ингибитора
протекают быстро и обратимы, так что
они существуют в равновесии. Позиции
этого равновесия будут зависеть от
концентраций реагентов. Учитывая, что
фермент участник обеих реакций их
равновесн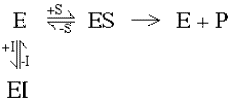 ые
состояния взаимосвязаны. Это означает,
что при высоких уровнях ингибитора
фактически все молекулы фермента будут
участвовать в образованииEIкомплекса, и фермент будет почти
полностью ингибирован. С другой стороны,
при высоких концентрациях субстрата
почти все молекулы фермента будут
связаны вESкомплексе и
ингибитор не сможет связаться с
ферментом. Высокие концентрации
субстрата снимают действие ингибитора.
Субстрат и ингибитор конкурируют
друг с другом.
ые
состояния взаимосвязаны. Это означает,
что при высоких уровнях ингибитора
фактически все молекулы фермента будут
участвовать в образованииEIкомплекса, и фермент будет почти
полностью ингибирован. С другой стороны,
при высоких концентрациях субстрата
почти все молекулы фермента будут
связаны вESкомплексе и
ингибитор не сможет связаться с
ферментом. Высокие концентрации
субстрата снимают действие ингибитора.
Субстрат и ингибитор конкурируют
друг с другом.
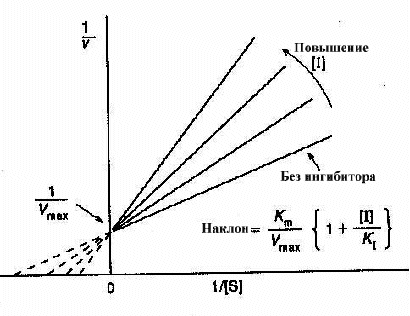
График Лаинуивера-Берка в случае классического конкурентного ингибирования
Эффект на KmКм - индикатор сродства субстрата и фермента. В присутствии конкурентного ингибитора некоторые молекулы фермента будут существовать как свободные ферменты, другие как комплексы ингибитора фермента. Первые будут иметь нормальное сродство, а вторые полностью неспособны к связыванию субстрата. Км измеряет полное сродство фермента в реагирующей смеси, которое будет представлять среднее значение между нормой и нулевым значением этого сродства, и поэтому будет явно меньше нормального значения. Так что конкурентный ингибитор уменьшает сродство субстрата и фермента, или увеличивает Км.
Эффект на Vmax Vmax - скорость при высоких концентрациях субстрата. Поскольку в этих условиях, ингибитор вытесняется субстратом, он не тормозит фермент вообще и, следовательно, конкурентные ингибиторы не замедляют реакцию при высоких концентрациях субстрата, и не изменяют Vmax. Это можно хорошо видеть на графике Лайнуивера-Берка. Наклон графика равенKm/Vmax. Увеличение наклона в присутствии ингибитора указывает на снижение скорости реакции при низких уровнях субстрата.
Наиболее часто приводимый пример конкурентного ингибирования - это использование малоновой кислоты для торможение дегидрогеназы янтарной кислоты. Наиболее близким структурным аналогом сукцината является малоновая кислота.
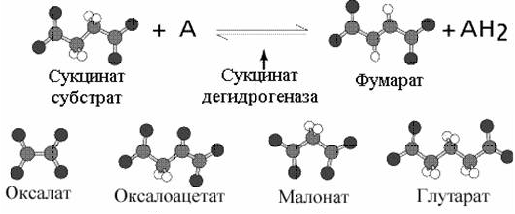
Примеры конкурентных ингибиторов.
Малоновая кислота тормозит активность дегидрогеназы янтарной кислоты, занимая активный центр на ферменте. Учитывая обратимость реакции, избыток янтарной кислоты снимет действие малоновой кислоты.
Принципы конкурентного торможения находят применение в медицинской практике.
Н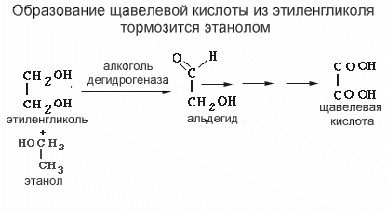 апример,
этиловый спирт используется с лечебной
целью как конкурентный ингибитор для
лечения отравлений этиленгликолем или
метиловым спиртом. В обоих случаях
проблема часто возникает среди
алкоголиков, потребляющих эти алкоголи
как дешевый заменитель этанола. Однако,
имеются другие ситуации, где может
возникать такое отравление. Производители
вина добавляют этиленгликоль к своим
винам, чтобы придать им внешний вид
более дорогих вин. В результате большое
количество ничего не подозревающих
людей могут потреблять это токсическое
соединение. Эти соединения часто
потребляются случайно взрослыми и
детьми. Животные, главным образом собаки
и коты, иногда вылизывают пролитый
антифриз из-за его особого вкуса.
апример,
этиловый спирт используется с лечебной
целью как конкурентный ингибитор для
лечения отравлений этиленгликолем или
метиловым спиртом. В обоих случаях
проблема часто возникает среди
алкоголиков, потребляющих эти алкоголи
как дешевый заменитель этанола. Однако,
имеются другие ситуации, где может
возникать такое отравление. Производители
вина добавляют этиленгликоль к своим
винам, чтобы придать им внешний вид
более дорогих вин. В результате большое
количество ничего не подозревающих
людей могут потреблять это токсическое
соединение. Эти соединения часто
потребляются случайно взрослыми и
детьми. Животные, главным образом собаки
и коты, иногда вылизывают пролитый
антифриз из-за его особого вкуса.
В таких случаях этиловый спирт - не яд, а продукт метаболизма. Алкогольдегидрогеназа катализирует образование соответствующего альдегида, который затем превращается в кислоту альдегиддегидрогеназой. Результатом этого процесса является образование формальдегида и муравьиной кислоты из метанола и глиоксаля и щавелевой кислоты из этиленгликоля. Альдегиды – основные токсические вещества , но и кислоты тоже дают нежеланные эффекты. Прежде всего, они могут вызывать ацидоз. Щавелевая кислота может кристаллизоваться в форме соли кальция в почечных канальцах, вызывая механическое повреждение.
Если затормозить эти реакции, метанол и/или этиленгликоль будут выделяться неизменяемыми. В ряде случаев это можно сделать, применяя конкурентный ингибитор. Этиловый спирт - такой ингибитор. Сродство алкогольдегидрогеназы к этиловому спирту примерно в 160 раз выше, чем к этиленгликолю. Задача врача поднять уровень этилового спирта в крови до 0.1 мг/l00 мл, что приведет к вытеснению этиленгликоля из реакции и выведению этиленгликоля с мочой.
С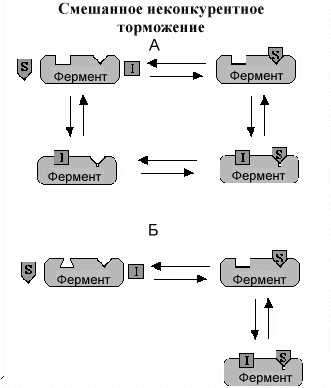 мешанные
неконкурентные ингибиторы
мешанные
неконкурентные ингибиторы
Неконкурентный ингибитор связываясь со специальным центром на ферменте, расположенным вдали от активного центра, вызывает конформационные изменения в активном центре. Это несколько напоминает один из типов конкурентных ингибиторов, но в отличие от этого механизма изменения конформации активного центра не препятствуют связыванию субстрата а тормозит проведение реакции и образование продукта..
Классический неконкурентный ингибитор не оказывает влияния на связывание субстрата. Изменения формы активного центра незначительны, хотя сродство к субстрату все же снижается. Такие ингибиторы часто называются смешанными ингибиторами, поскольку они проявляют свойства конкурентных и неконкурентных типов. Фактически классические неконкурентные ингибиторы встречаются очень редко.
Кинетика смешанных неконкурентных ингибиторов
Фермент может связываться и с субстратом и с ингибитором с образованием комплексов:
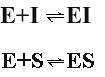
Однако в отличие от конкурентного торможения и субстрат ингибитор способны одновременно связываться с ферментом, образуя тройные комплексы:
Т акой
комплекс не способен катализировать
реакцию.
акой
комплекс не способен катализировать
реакцию.
О![]() братите
внимание, что комплексEISможет возникать двумя путями, но конечный
результат один и тот же - образуется
неактивный фермент. Ингибитор может
работать одинаково хорошо при низких
концентрациях субстрата, когда большая
часть молекул фермента находится в
форме свободного фермента, и при высоких
концентрациях субстрата, когда фермент
находится в формеESкомплекса, поскольку ингибитор
связывается одинаково хорошо с обоими
из них.
братите
внимание, что комплексEISможет возникать двумя путями, но конечный
результат один и тот же - образуется
неактивный фермент. Ингибитор может
работать одинаково хорошо при низких
концентрациях субстрата, когда большая
часть молекул фермента находится в
форме свободного фермента, и при высоких
концентрациях субстрата, когда фермент
находится в формеESкомплекса, поскольку ингибитор
связывается одинаково хорошо с обоими
из них.
Влияние на Km Классический смешанный неконкурентный ингибитор не оказывает влияния на связывание субстрата, сродство фермента к субстрату и следовательно не влияет на Км.
Смешанный ингибитор позволяет субстрату связываться, но снижает его сродство, так что Км повышается.
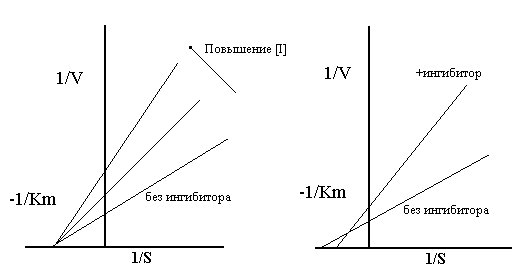
График Лаинуивера-Берка для неконкурентного классического(слева) и смешанного типов ингибиторов
Влияние на Vmax Неконкурентные и классический и смешанный варианты, тормозят активность при высоких концентрациях субстрата, так чтоVmaxснижается.
Влияние на Vmax/Km Неконкурентные ингибиторы могут работать при низких концентрациях субстрата так, чтоVmax/Km снижается.
Г![]() рафик
Лайнуивера-Берка для типичного
классического смешанного неконкурентного
ингибитора и для смешанного неконкурентного.
На графике для смешанного конкурентного
ингибитора две линии по разному
пересекают 1/ [S], указывая
на увеличение Км, хотя на графике для
классического смешанного неконкурентного
ингибитора, значенияKmидентичны. Однако оба графика показывают
увеличение наклона в пересечении на
оси 1/V, демонстрируя, что
ингибитор является эффективным при
высоких и низких концентрациях фермента.
рафик
Лайнуивера-Берка для типичного
классического смешанного неконкурентного
ингибитора и для смешанного неконкурентного.
На графике для смешанного конкурентного
ингибитора две линии по разному
пересекают 1/ [S], указывая
на увеличение Км, хотя на графике для
классического смешанного неконкурентного
ингибитора, значенияKmидентичны. Однако оба графика показывают
увеличение наклона в пересечении на
оси 1/V, демонстрируя, что
ингибитор является эффективным при
высоких и низких концентрациях фермента.
Заключение. Смешанные неконкурентные ингибиторы связываются с центром, который является отдаленным от активного центра. Они тормозят активность фермента, вызывая изменения конформации , препятствующие превращению субстрата в продукт. Субстрат и ингибитор способны к связыванию с ферментом одновременно, образуя тройной комплекс. Это означает, что ингибитор не конкурирует с субстратом при высоких концентрациях субстрата и работает одинаково хорошо и при низких и высоких концентрациях субстрата. Правда способность связываться (сродство) с комплексом ингибитор+фермент у субстрата может снижаться, хотя на практике нет особых различий между так называемым смешанным ингибитором и классическим неконкурентным ингибитором.
Неконкурентные ингибиторы не могут связаться со свободным ферментом.
Понятие неконкурентный ингибитор часто смешивают с понятием смешанный неконкурентный ингибитор! Ингибиторы такого типа встречаются редко, хотя они имеют прямое отношение к изучению мультисубстратных ферментов.
Главная особенность этих ингибиторов - они не способны связываться со свободным ферментом. Они связываются только с фермент-субстратным комплексом. Это объясняется тем, что субстрат сам непосредственно включен в связывание ингибитора или связывание субстрата вызывает изменение конформации в связывающем ингибитор центре, облегчающее связывание ингибитора. Связывание ингибитора вызывает торможение активности фермента и превращение субстрата в продукт. Это может быть следствием прямого взаимодействия, или изменения конформации активного центра.
Неконкурентных ингибиторы неактивны при низких концентрациях субстрата.
Д![]() ля
связывания неконкурентного ингибитора,
фермент должен сначала связаться с
субстратом:
ля
связывания неконкурентного ингибитора,
фермент должен сначала связаться с
субстратом:
![]() и
только затем происходит связывание с
ингибитором:
и
только затем происходит связывание с
ингибитором:
К![]() омбинация
ингибитора со свободным ферментом:
омбинация
ингибитора со свободным ферментом:
невозможна.
Это означает, что неконкурентные ингибиторы неактивны при очень низких концентрациях субстрата, когда почти весь фермент присутствует в свободной форме. При высоких концентрациях субстрата, когда большая часть молекул фермента присутствует в форме комплекса с субстратом, ингибитор эффективен.
Влияние на Km Неконкурентные ингибиторы оказывают довольно неожиданный эффект на Км. Поскольку ингибитор связывается с комплексом субстрата и фермента, он эффективно уменьшает концентрацию этого комплекса, превращая часть его в тройнойEIS. По закону действующих масс это способствует смещению равновесия субстрат связывающей реакции вправо и тем самым ингибитор увеличивает количество субстрата, который связывается с ферментом, давая очевидное увеличение сродства фермента к субстрату и уменьшение Км.
Эффект на Vmax Поскольку ингибитор не вытесняется большими количествами субстрата, а наоборот, при больших количествах субстрата быстрее идет образования его комплекса с ферментом, необходимого для проявления действия ингибитора и поэтому уменьшаетсяVmax.
Влияние на Vmax/Km Так как эти ингибиторы не работают при низких концентрациях субстрата,Vmax/Km не меняется.
Типичный график Лаинуивера - Берка для неконкурентного ингибитора представлен на рис. 2-12.
Вывод. Неконкурентные ингибиторы могут связываться только с комплексом субстрат-фермент, но не со свободным ферментом. В результате они не действуют при очень низких концентрациях субстрата. Они способствуют явному увеличению сродства фермента к субстрату, поскольку большее количество субстрата связывается с ферментом для образования, правда, неактивного тройного комплекса
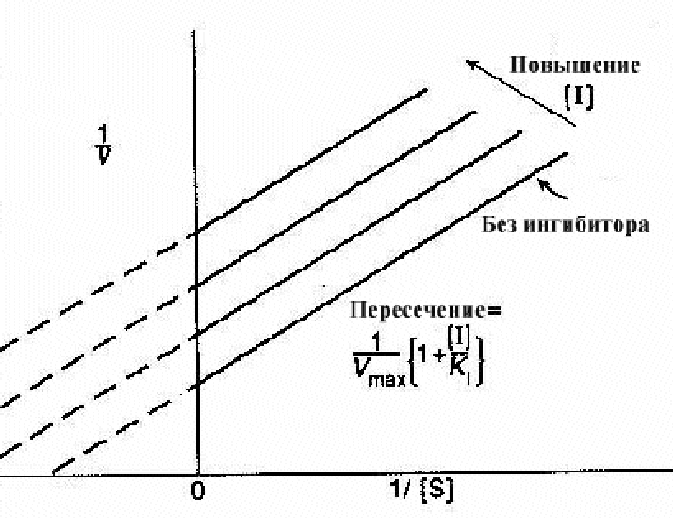
Рис 2-12. График Лаинуивера-Берка для неконкурентных ингибиторов
Торможение продуктом реакции- пример конкурентного торможения.
Заключительный этап в действии любого фермента -выделение продукта. Это важный этап в работе фермента. Продукт связан с активным центром теми же связями, которые связывали и субстрат. Это взаимодействие похоже на субстрат связывающую реакцию, и подобно этой реакции, чаще всего является очень быстрой и легко обратимой. В результате этого, молекулы продукта способны к связыванию со свободным ферментом, образуя комплекс продукта и фермента. Как и в случае действия конкурентного ингибитора, продукт и субстрат пытаются занять активный центр, но не могут связываться одновременно. Поэтому предшествующее связывание продукта предохраняет фермент от связывания с субстратом , и продукт эффективно действует как ингибитор. В ферменте, катализирующем реакцию первого порядка это специализированная форма конкурентного торможения, поскольку субстрат и продукт конкурируют друг с другом. В ферментах с больше чем одним субстратом это выглядит более сложно.
В ферментных кинетических исследованиях мы почти всегда изучаем начальные скорости, которые является скоростью реакции, начинающейся немедленно после добавления фермента к субстрату. В это период времени, конечно, продукта нет, поскольку он еще не успел возникнуть, так что мы обычно игнорируем торможение продуктом в наших кинетических исследованиях.
Субстрат может быть ингибитором фермента
О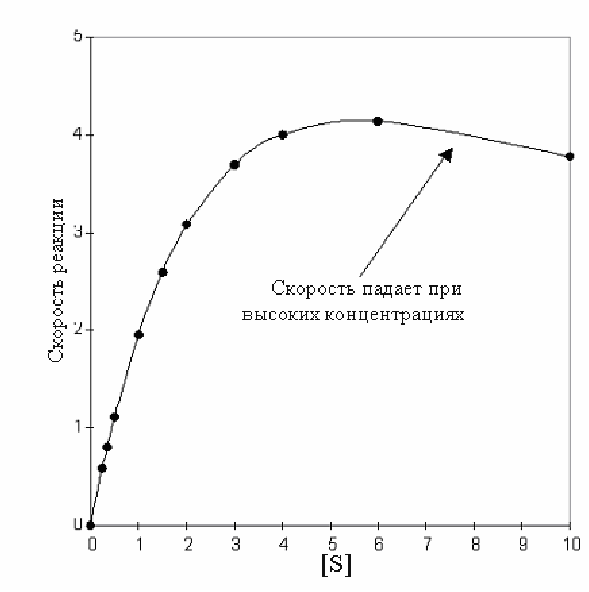 бычно
увеличение концентрации субстрата
увеличивает скорость реакции фермента.
Однако на некоторые ферменты большие
количества субстрата могут оказывать
противоположный эффект и фактически
замедлять реакцию. Примером может
служить инвертаза или-
фруктофуранозидаза (КФ.3.2.1.26), который
катализирует реакцию гидролиза
бычно
увеличение концентрации субстрата
увеличивает скорость реакции фермента.
Однако на некоторые ферменты большие
количества субстрата могут оказывать
противоположный эффект и фактически
замедлять реакцию. Примером может
служить инвертаза или-
фруктофуранозидаза (КФ.3.2.1.26), который
катализирует реакцию гидролиза
Рис 2-13. Влияние концентрации субстрата на скорость реакции
сахарозы на составляющие моносахариды глюкозу и фруктозу.
Предполагают, что причиной торможения может быть попытка двух молекул субстрата одновременно связаться с активным центром и пока молекулы субстрата связаны с активным центром фермент неактивен. Для такого связывания необходимо, чтобы вторая молекула субстрата «вставилась» в активный центр быстро вслед за первой, пока последняя не успела занять полноценную позицию для катализа. Поскольку столкновения между ферментом и субстратом полностью случайны, такое может случиться только при высоких концентрациях субстрата, когда частота случайных столкновений увеличивается, и явление торможения может быть замечено исследователем. Пример такого влияния показан на рис 2-13.
Т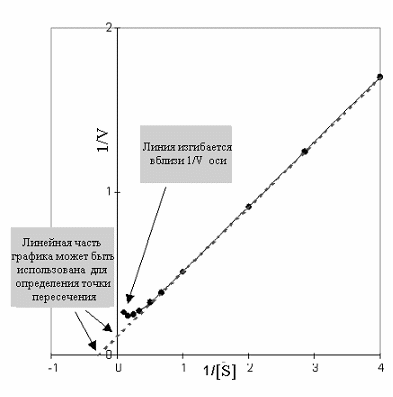 ак
как торможение субстратом происходит
при высоком уровне субстрата, начальная
часть (низкая концентрация субстрата)
этого графика идентична таковой для
нормального
ак
как торможение субстратом происходит
при высоком уровне субстрата, начальная
часть (низкая концентрация субстрата)
этого графика идентична таковой для
нормального
Рис 2-14. График Лаинуивера-Берка по данным рис 2-13.
фермента. При высоком уровне субстрата кривая снижается, что свидетельствует о торможении скорости реакции.
Так как фермент не следует предписаниям уравнения Михаэлиса и Ментен (не прямоугольная гипербола), график Лайнуивера Берка также не линеен. Поэтому вблизи оси 1/Vна участке высокой концентрации субстрата, линия изгибается вверх, указывая на снижение скорости. Продолжая линейную часть графика до пересечения с осями можно определить основные кинетические параметры обычным способом.
Табл 2- 5. Эффекты обратимых ингибиторов на кинетические константы
|
Тип ингибитора |
Эффект |
|
Конкурентный (Iсвязывается только кE) |
Увеличение Km,аVmaxостается без изменений |
|
Неконкурентный (Iсвязывается только сES) |
Снижена VmaxиKmОтношениеVmax.Kmостается без изменений |
|
Смешанный неконкурентный (Iсвязывается сEилиES) |
Снижается Vmax .Kmостается без изменений |
Регуляция активности ферментов путем химической модификации: Реакции ограниченного протеолиза, аденилирования, рибозилирования, ацетилирования, фосфорилирования (роль гормонов, АЦ-комплекса, цАМФ, цГМФ, ионов Ca).
Активность фермента можно изменить путем ковалентной модификации его структуры.
Ковалентная модификация структуры ферментов может быть обратимой и необратимой.
А. Необратимые изменения активности фермента. Существует большое число реакций, катализируемых протеолитическими ферментами. Как правило, такие ферменты образуются в форме неактивных предшественников (проферментов), которые затем активируются путем действия других протеаз. Схеме активирования химотрипсина показана ниже на рис.2-15.
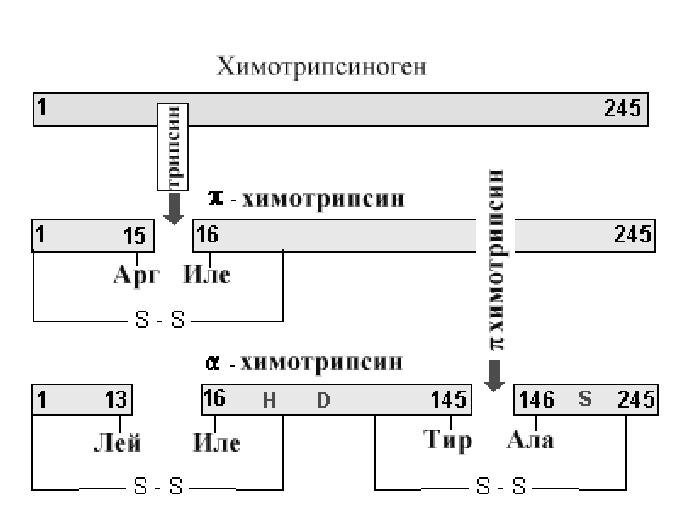
Рис 2-15. Схема протеолиза молекулы химотрипсиногена. Вначале под влиянием трипсина происходит отщепление полипептида из 15 аминокислот с образованием активного -химотрипсина, который путем аутокатализа превращается в-химотрипсин
Ниже в таблице приводятся примеры других ферментов, которые подвергаются активируются путем протеолиза
|
Фермент |
Профермент |
Место образования |
|
Трипсин |
трипсиноген |
поджелудочная железа |
|
Химотрипсин |
химотрипсиноген |
поджелудочная железа |
|
Эластаза |
проэластаза |
поджелудочная железа |
|
Карбоксипептидаза |
прокарбоксипептидаза |
поджелудочная железа |
|
Фосфолипаза А2 |
профосфолипаза А2 |
поджелудочная железа |
|
Пепсин |
пепсиноген |
желудок |
|
Тромбин |
протромбин |
свертывание крови |
|
C1r |
C1r- |
комплемент |
|
Хитин синтаза |
зимоген |
стенка клеток дрожжей |
В качестве примера можно рассмотреть ферменты поджелудочной железы. Эти ферменты синтезируются в форме проферментов клетками поджелудочной железы и сохраняются в них в составе гранул проферментов. Высвобождение проферментов регулируется гормонами холецистокинином, панкрезимином и секретином. Усвоение белков требует скоординированного действия различных ферментов. Трипсиноген активируется энтеропептидазой выделяемой клетками щеточной каймы кишечника. Энтеропептидаза катализирует удаление гексапептида (Вал Асп Асп Асп Асп Лиз) от Nконца трипсиногена. Задача системы регуляции в данном случае обеспечить активирование фермента в месте его действия. Если активирование произойдет в месте синтеза, то это повлечет за собой разрушение самой железы (острый панкреатит). Небольшое количество энтеропептидазы, необходимое для активирования трипсиногена обеспечит последующее усиление (амплификацию) сигнала за счет образования больших количеств активного трипсина, который в свою очередь активирует другие протеазы кишечника. Такие цепные реакции усиления характерны для многих протеолитических систем организма (свертывание крови, система комплемента и т.д.) Процесс останавливается путем протеолиза самих ферментов Средняя ежедневная продукция короткоживущих гидролитических ферментов поджелудочной железой - около 10 г. Протеолитические ферменты широко распространены в клетках и это могло создать значительные проблемы, если бы не специальные белковые игибиторы, присутствующие в тканях и крови.
Б. Обратимые изменения активности фермента путем ковалентной модификации.
Фосфорилирование ферментов (рис 2-16) - одна из наиболее популярных реакций, позволяющая при присоединении остатка фосфорной кислоты изменить конформацию фермента и перевести фермент из активного состояние в неактивное или наоборот. Этот механизм ковалентной модификации обеспечивается киназами (присоединение остатков фосфорной кислоты за счет АТФ) и фосфатазами (гидролитическое удаление остатков фосфорной кислоты).
Более подробно об этих механизмах будет рассказано в разделах, посвященных механизму действия гормонов.
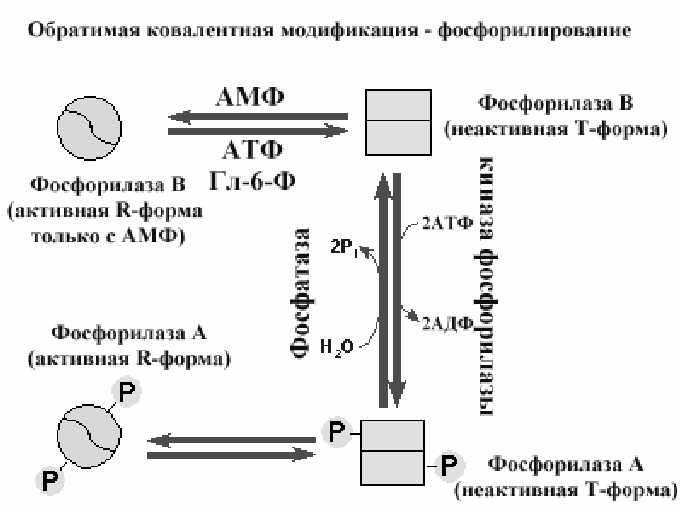
Рис 2-16. Обратимая ковалентная модификация путем фосфорилирования (киназа фосфорилазы) и дефосфорилирования (фосфатаза) фосфорилазы.
Регуляция активности с помощью гормонов.
Гормональная регуляция осуществляется на генетическом уровне путем обратного фосфорилирования. Например под действием адреналина и глюкагона происходит активация процесса распада гликогена, в ходе этого процесса образуется небелковое соединение - цАМФ, цАМФ - внутриклеточный гормон (вторичный посредник) яв-ся аллостерическим регулятором большого числа протеинкиназ. цАМФ образуется из АТФ под действием аденилатциклазы:
Гормон, циркулирующий в крови, попадает в межклеточную жидкость и контактирует с поверхностью клетки, где расположены рецепторы (Rs и Ri), белки узнающие и связывающие гормон.
Гормональный сигнал поступает на АЦ через белки посредники (Gs и Gi), которые активируются в условиях присоединения ГТФ. Уровень, образовавшийся под действием АЦ, цАМФ определяется не только активностью АЦ, но и активностью фосфодиэстераз, которые циклизируют цАМФ до АМФ.
Кроме цАМФ, существуют цГМФ, цУМФ, цЦМФ. Наибольшее значение имеет цГМФ. Она образуется под действием гуанилатциклазы, расположенной как в наружной мембране, так и внутри клетки, цГМФ единственный фермент, который реагирует на концентрацию Н2О2и активируется под действием продуктов перекисного окисления.
цГМФ оказывает эффекты противоположные цАМФ.
цАМФ находится в тесном контакте с ионами Ca2+: высокая концентрация цАМФ в возбудимых тканях приводит к высокой [Ca2+] и стимуляции клетки. И наоборот, резкое уменьшение [Ca2+] тормозит АЦ и снижает уровень цАМФ.
Изоферменты, их природа, биологическая роль, строение ЛДГ.
Изоферменты- это группа родственных ферментов, катализирующих одну и ту же реакцию. Они происходят из одного предшественника за счет дупликациии гена с последующей мутацией образуемых аллелей. Они отличаются между собой:
1) скорстью катализа;
2) направлением катализируемой реакции;
3) условиями протекания реакции;
4) чувствительностью к регуляторам, факторам среды. (Более или менее устойчивы к ингибиторам);
5) сродством к субстрату;
6) особенностями структуры молекулы, ее ИЭТ, Mr, размерами и зарядом.
Изоферменты имеют адаптивное значение, т. е. придают специфику метаболизма.
Изоферменты обеспечивают межорганную связь, например, в процессе мышечной деятельности.
В миокарде и печени существуют различные изоферменты ЛДГ, которые обеспечивают метаболизм лактата:
ЛДГ4,5
в печени: ПВК -----> лактат
ЛДГ1,2
в сердце: лактат ------> ПВК
ЛДГ - олигомерный фермент, состоящий из 4-х субъединиц 2 типов.
H (heart) и M (muscle).
Существует 5 изоферментных форм:
HHHH HHHM HHMM HMMM MMMM
H4 H3M H2M2 HM3 M4
ЛДГ1, ЛДГ2, ЛДГ3, ЛДГ4, ЛДГ5.
Поскольку H-протомеры несут более выраженный отрицательный заряд, то изофермент H4 (ЛДГ1) будет мигрировать при электрофорезе с наибольшей скоростью к аноду.
С наименьшей скоростью к аноду будет двигаться М4.
Остальные изоферменты занимают промежуточное положение.
Изоферменты ЛДГ локализованы в различных тканях:
ЛДГ1,2 ----> мозг, аэробные ткани (миокард).
ЛДГ3 ----> лейкозные клетки.
ЛДГ4,5 ----> анэробные ткани: мышечная, скелетная.
Изоферменты появляются на различных этапах онтогенеза и реализуют программу индивидуального развития.
Изоферментный профиль меняется в процессе развития.
При патологиях имеется существенный изоферментный сдвиг.
Изменение активности ферментов в онтогенезе.
Изменение активности в онтогенезе.
Онтогенез человека развивается по определенной генетической программе, которая записана на уровне ткани, всего организма в гипоталамусе.
1. Внутриутробный период.
Характеризуется высокой активностью ферментов синтеза белка, липидов, происходит увеличение массы организма. Плод находится в анаэробных условиях и для метаболизма характерно анаэробная направленность.
Основной источник энергии - жирные кислоты, поступающие из организма матери; ЖК также выполняют строительную функцию (фосфолипиды мембран).
Глюкоза утилизируется анаэробным путем (анаэробный гликолиз), т. к. ткани плода не способны к ГНГ, и идет на развитие ЦНС.
2. Пренатальный период.
Характеризуется изменением активности ферментов, происходит подготовка организма к пребыванию в аэробной среде. Изменяется спектр гемоглобина, уменьшается его сродство к кислороду, изменяется активность митохондриальных ферментов.
3. Грудной
Потребность в глюкозе резко возрастает, она начинает утилизироваться аэробно, но примерно до двух лет основным источником энергии является все же липиды, причиной чего является соматотропин. (Гормон роста).
4. Ранний дошкольный период.
С 3-х до 5-и лет. В этот период клетки начинают питаться углеводами. Происходит стабилизация обмена и интенсивная миелизация нервных волокон.
5. Школьный и пубертантный период.
Обмен веществ модулируется под действием половых гормонов.
6. Зрелый.
Происходит стабилизация массы тела, репродуктивного гомеостаза. После 35-40 лет основным источником энергии являются опять липиды, что связано с ослаблением чувствительности тканей к Гл и изменение гормонального фона: гиперстресс (увеличивается уровень гормонов) заставляет клетку работать на пределе, т. е. использовать в качестве энергии жиры.
|
Заведующий кафедрой биологической химии, д.м.н., проф. |
Грицук А. И. |
___________ |
21.10.2006
Министерство здравоохранения Республики Беларусь
УО «Гомельский государственный медицинский университет»
Кафедра биологической химии
Обсуждено на заседании кафедры (МК или ЦУНМС)
Протокол № _________________200__года
ЛЕКЦИЯ по биологической химии
наименование дисциплины
для студентов _2__ курсалечебногофакультета
Тема Ферменты 4. Номенклатура и классификация.
Время 90 мин.
Учебные и воспитательные цели:
Дать представление:
О локализации ферментов в клетке, органоспецифических и маркерных ферментах.
О качественном обнаружении и количественном определении активности. О единицах активности (МE, катал), удельной активности, числе оборотов ферментов.
О сопряженных ферментных системах и их применении. О номенклатуре, классификации ферментов (тривиальной, рациональной, систематической). О принципах классификации.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф.Биологическая химия. М.: Медицина, 1990. С. 126–132; 1998. С. 157–168.
Николаев А. Я.Биологическая химия. М.: Высшая школа, 1989. С. 52–92.
Дополнительная
Марри Р. и др. Биохимия человека. М.: Мир, 1993. Т. 1. С. 63–75.
Филиппович Ю. Б. Основы биохимии. М.: Высшая школа, 1993. С. 105–144.
Врожденные и приобретенные энзимопатии / Под ред. Ташева Т. М.: Медицина, 1980.
Вилкинсон Д. Принципы и методы диагностической энзимологии. М.: Медицина, 1981.
Руководство по клинической лабораторной диагностике. Киев: Вища школа, 1990. С. 167–186.
Зилва Ф., Пеннел Дж. Клиническая химия в диагностике и лечении. М.: Медицина, 1986. С. 372–388.
МАТЕРИАЛЬНОЕ ОБЕСПЕЧЕНИЕ
1. Мультимедийная презентация.
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№ п/п |
Перечень учебных вопросов |
Количество выделяемого времени в минутах |
|
|
Локализация ферментов в клетке, органоспецифические и маркерные ферменты. |
30 |
|
|
Качественное обнаружение и количественное определение активности. Единицы активности (МE, катал). Удельная активность. Число оборотов ферментов. |
30 |
|
|
Сопряженные ферментные системы их применение. Номенклатура, классификация ферментов (тривиальная, рациональная, систематическая). Принципы классификации. |
30 |
Всего 90 мин
Локализация ферментов в клетке, органоспецифические и маркерные ферменты.
Все ферменты и метаболические процессы компартментализованы (разделены и изолированы).
В нормальной клетке находится более 1000 ферментов.
Упорядоченное взаимодействие ферментов достигается путем многоуровневой регуляции и компартментализации.
Зная локализацию ферментов в клетке и определяя их активность в крови, можно судить о степени деструкции ткани.
Ядро: локализованы РНК-полимеразы, НАД-синтетаза, ферменты, участвующие в репликации ДНК.
Митохондрии: ферменты тканевого дыхания, окислительного фосфорилирования, ферменты b-окисления ЖК, цикла Кребса, пируватдегидрогеназного комплекса, синтеза мочевины.
Лизосомы: гидролитические ферменты с оптимумом рН в области 5 (пептидазы, эстеразы, нуклеазы).
Рибосомы: ферменты белкового синтеза.
ЭПС: ферменты синтеза липидов, ферменты гидроксилирования, детоксикации (метилирования, ацетилирования), конъюгации.
Мембраны: Na-K-АТФаза, аденилатциклаза, ферменты транспорта субстратов.
Цитоплазма: ферменты гликолиза, ПЦ, активации а/к, ферменты синтеза жирных кислот, ГНГ.
Мультиферментные системы локализуются в структуре органелл таким образом, что каждый фермент располагается в непосредственной близости от следующего фермента данной последовательной реакции.
Благодаря также компартментализации в клетке могут одновременно протекать 2 несовместимых процесса, например: b-окисление ЖК (в митохондриях) и синтез ЖК (в цитоплазме).
Органоспецифические ферменты:
Под органоспецифичностью понимают наличие метаболических путей, присущих только данному органу.
Так вот органоспецифические ферменты - это ферменты, катализирующие определенные метаболические пути, присущие определенному органу.
Хотя органы и имеют различное выражение того или иного метаболического пути, они имеют важное значение для диагностики многих заболеваний, путем определения их активности:
так для печени характерна высокая активность АсАТ, АлАТ, сорбитдегидрогеназы, ГДГ. Причем активность АлАТ выше, чем АсАТ, т. к. АсАТ лучше спрятана во внутриклеточных структурах.
Костная ткань - щелочная фосфатаза.
Простата - кислая фосфатаза.
Glandula parotis et pancreas - амилаза.
Миокард - ЛДГ1 и ЛДГ2 , креатинкиназа (ММ и MB-изоферменты).
Мышцы - ЛДГ4,5; ММ.
При нарушении целостности тканей этих органов, ферменты выделяются в сыворотку крови, где их активность резко повышается. В зависимости от того, активность какого фермента повысилась можно судить не только локализации каталитического процесса, но и о степени его тяжести.
Но для более конкретной и точной диагностики заболеваний, для определения интенсивности и глубины повреждения тканей нужны маркерные ферменты. Это ферменты, принадлежащие определенной конкретной органелле:
сукцинатдегидрогеназа - внутренняя мембрана митохондрий;
кислые гидролазы - лизосомы;
ферменты гликолиза - цитоплазма и т. д.
Качественное обнаружение и количественное определение активности. Единицы активности (МE, катал). Удельная активность. Число оборотов ферментов.
О скорости ферментативной реакции судят или по скорости убыли субстрата, или по скорости образования продукта.
За единицу активности любого фермента (Е) принимается то количество фермента которое в оптимальных условиях катализирует превращение 1 мкмоля субстрата в 1 мин.
Существует другая единица активности:
1 катал - количество фермента, который катализирует превращения 1 моля субстрата в 1 секунду.
1 Е фермента = 16,67 нкатал.
Для выражения активности фермента пользуются определением удельной и молекулярной активности.
Удельная активность - число Е ферментативной активности в расчете на 1 мг белка.
Чем выше очистка фермента, тем выше удельная активность.
Число оборотов фермента (молекулярная активность) - число молекул S,подвергающихся превращению одной молекулой фермента в 1 минуту. (Или число элементарных актов катализа, осуществляемой 1 молекулой фермента за 1 минуту).
Число оборотов широко варьирует, например:
1. Карбоангидраза (катализирует перенос Н2СО3) совершает 36 000 000 оборотов.
2. Каталаза - 4000 оборотов.
3. Фосфоглюкомутаза - 1240 об/мин.
Для качественного обнаружения и количественного определения активности сложных ферментов используют следующие методы: ЛДГ CH3CH3
лактат -------- ПВК CH-ОН ------- С=О
НАД НАД.Н+Н COOH COOH
НАД.Н интенсивно поглощает свет ( =340 нм). По изменению ПВК можно судить о НАДН.
На одну молекулу лактата образуется одна молекула НАД.Н.
НАД.Н интенсивно флюоресцирует. Интенсивность флюоресценции будет пропорциональна концентрации.
В общем виде под активностью понимают количество фермента или биологического материла, содержащего фермент, которое при определенных условиях катализирует в единицу времени превращение определенного количества реагента, называемого субстратом. Активность - это изменение количества субстрата под влиянием фермента в единицу времени. Под изменением субстрата понимают снижающееся в единицу времени количество субстрата или же увеличивающееся количество продукта. Понятие "активность фермента" по сути дела идентична понятию "скорость ферментативной" реакции. Ферментативная активность выражается в единицах активности. В связи с существованием различных систем единиц исчисления введена интернациональная (стандартная) единица активности. Она носит символ "U" (unit-единица) и определяется как 1 мкмоль субстрата/мин. В системе СИв качестве единицы ферментативной активности используют "катал" (kat). Катал определяется как 1 моль/сек.
1kat = 1 моль/сек.
Размерность её слишком велика, на практике пользуются меньшими кратными значениями, начиная с нанокатала (нкат). Это одна миллиардная катала или 10-9кат. В сравнении с международной единицей следующее уравнение
1 U = 16,67 нкат
В практике лабораторий широко пользуются понятием удельная активность. Для этого числоcтандартных единиц пересчитывают на какую-либо единицу сравнения. Это может быть мг белка в пробе или объем исследуемой биологической жидкости. Определение активности ферментов широко распространено в любой современной клинической лаборатории.
При исследовании кинетики реакций используется и такое понятие как молекулярная активность. Она показывает, сколько молекул субстрата в секунду превращаются в продукт 1 молекулой фермента и используется для сравнительной характеристики активности нескольких ферментов.
Пример вычисления активности фермента:
|
Исходные данные: |
Через 10 мин: |
|
25.0 x10-3моль л--1пептида-субстрата, объем реакционной смеси 2.5 мл, 0.50 µг химотрипсина1 |
18.6 x10-3моль л--1пептида -субстрата, Объем реакционной смеси 2.5 мл, 0.50 µг химотрипсина. |
|
Использованный субстрат |
= 6.4 x10-3моль л-1за 10 мин |
|
Скорость реакции |
= 6.4 x10-4моль л-1мин-1 |
|
Активность Фермента(скоростьxобъем) |
= 6.4 x10-4моль л-1мин-1x2.5x10-3л = = 1.6x10-6моль мин-1 |
|
Удельная активность(активность / масса) |
= 1.6 x10-6моль мин-1/ 0.50 µг = = 3.2x10-6моль µг-1мин-1 |
|
Число оборотов(уд. акт.xмолярная масса) |
= 3.2 x10-6моль µг-1мин-1x25,000x106µг моль-1= 8.0x104мин-1=1330 сек-1 |
Если удельная активность, рассчитанная выше, относится к чистому химотрипсину, образец, давший, например, удельную активность 2.0 x10-7моль µг-1мин-1- 100 %x2.0x10-7/ 3.2x10-6или 6.3 % чистоты. 1.0 µг такого образца на самом деле содержит лишь 0.063 µг химотрипсина и 0.937 µг примесей.

Рис2-4. Молярное поглощение НАД+,НАДН+Н+, ФАД, ФАДН2 при разных длинах волн поглощаемого света
Методы исследования активностиопределяются механизмом реакции и природой определяемого вещества. Наиболее широко используются:
Измерение изменения спектральных свойств (измерение поглощения света в видимой или ультрафиолетовой области, измерение флюоресценции) при помощи спектрофотометров, ФЭКов, спектрофлуориметров. Эти методы применяют и для определения количества продуктов или субстратов реакции, и для изменений количества коферментов, участвующих в реакции. Последнее нашло широкое применение в практике клинических биохимических лабораторий. В основе этих методов лежит закон Beer-Lambert:A= xcxl =log(I0/I) (, поглощение 1Mраствора вещества при специфической длине волны или молярный коэффициент экстинкции;c, концентрация ;A, поглощение ;l, длина в см кюветы спектрофотометра ;I0, интенсивность падающего света;I, интенсивность прошедшего света). В случае, если молярный коэффициент экстинкции ( исследуемого вещества неизвестен, исследователь определяет экспериментально зависимость между поглощением света исследуемого раствора и концентрацией этого вещества и использует полученную закономерность в форме стандартного (калибровочного) графика.
На рисунке 2-4 показаны спектральные характеристики коферментов НАД и ФАД в окисленной и восстановленной форме. Измерение поглощения при 340 нм используется для количественной оценки активности ферментов, катализирующих окислительно-восстановительные реакции cучастием НАД. Вот пример такого расчета для реакции, катализируемой лактатдегидрогеназой В этой реакции молочная кислота окисляется, передавая водороды на НАД+. При этом НАД+восстанавливается до НАДН +Н+., который в отличие от НАД+поглощает свет с длиной волны 340 нм. Допустим, за время проведения реакции поглощение при длине волны 340 нм изменялось на 0.31 единицы в минуту. Измерения проводили в кювете шириной 1 см. Коэффициент молярной экстинкции для НАДН при 340 нм= 6200 л моль-1см-1.
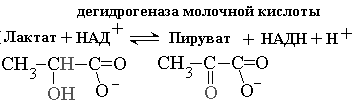
|
Увеличение [НАДH] = |
Увеличение поглощения e.l |
0.31 6200 |
=5.0 х10-5 моль/л |
Эту величину можно использовать для оценки скорости реакции.
Измерение изменений концентрации высвобождаемых или поглощаемых во время реакции H+или ОН-при помощиpH-стата (устройство, которое автоматически добавляет кислоту или основание, сохраняя постоянствоpHв реагирующей смеси)
Химический анализ с использованием высокоразрешающей жидкостной или газовой хроматографии, или ЯМР или тонкослойной хроматографии. (АТФазы)
Изотопный анализ (например, с использованием радиоактивного 32P)
Сопряженные ферментные системы их применение. Номенклатура, классификация ферментов (тривиальная, рациональная, систематическая). Принципы классификации.
С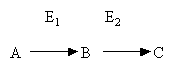 опряженные
реакции – используются в случаях, если
нет возможности прямо определить
количество продукта исследуемой
реакции. В таких случаях в реагирующую
смесь добавляется фермент (Е2)
катализирующий превращение образующегося
продукта в реакции, которую можно
оценить количественно, одним из
вышеперечисленных методов.
опряженные
реакции – используются в случаях, если
нет возможности прямо определить
количество продукта исследуемой
реакции. В таких случаях в реагирующую
смесь добавляется фермент (Е2)
катализирующий превращение образующегося
продукта в реакции, которую можно
оценить количественно, одним из
вышеперечисленных методов.
Если фермент Е2присутствует в избытке, скорость образованияCотражает скорость образования В.
Например, сопряженное исследование активности глюкокиназы (используется избыток глюкозо-6-фосфат дегидрогеназы и НАДФ+)
Глюкоза + AТФ → глюкоза 6-Ф +AДФ : (катализируется глюкокиназой –Е1) Глюкоза-6-Ф + НАДФ+ → 6-фосфоглюконолактон + НАДФН +H+ : ( катализируется глюкоза-6Ф –дегидрогеназой – Е2):
Скорость образования НАДФH(измеряется по поглощению при 340 нм) пропорциональна активности глюкокиназы (см выше)
В настоящее время известны и используются 3 вида классификации ферментов:
1. Тривиальная (исторически сложившаяся) номенклатура: (пепсин, трипсин).
2. Рациональная предложена французским физиологом П. Дюкло в 1883 году (к корню названия субстрата прибавляется суффикс
- аза (липид - липаза, протеин - протеаза и т.д.).
3. Современная классификация рассмотрена и утверждена V Всемирным биохимическим конгрессом в г. Москве в 1961 г. В основу ее положен тип катализируемой реакции (всего 6 классов):
1) Оксидоредуктазы: катализируют окислительно-восстановительные реакции, лежащие в основе биологического окисления. Название дается по схеме: донор: « акцептор-оксидоредуктаза» ---> лактат: НАД-оксидоредуктаза.
Различают аэробные дегидрогеназы или оксидазы, катализирующие перенос протонов (е) непосредственно на кислород; анаэробные дегидрогеназы ускоряющие перенос протонов (е) на промежуточный S, но не на кислород; цитохромы - катализируют перенос только е. Сюда также относятся каталаза и пероксидаза.
2) Трансферазы: ферменты, катализирующие перенос (внутри- и межмолекулярный) различных групп атомов. Название дается по форме: «донор - транспортируемая группа - трансфераза ---> метил-, формилтрансферазы, аминотрансферазы.
Оба этих класса ферментов работают при участии коферментов, которые являются водорастворимыми витаминами: В6, В12, В1, В15.
3) Гидролазы: ферменты, катализирующие расщепление внутримолекулярных связей при участии молекулы воды.
Название: «субстрат-гидролаза». К ним относятся все ферменты ЖКТ; в частности: эстеразы - гидролиз сложных эфиров; гликозидазы - гидролиз гликозидных связей углеводов; пептидгидролазы - гидролиз пептидных связей.
4) Лиазы - ферменты, расщепляющие C-C, C-N, C-O связи не гидролитическим путем с образованием двойной связи. Название: «субстрат-лиаза». Они обеспечивают отщепление CO2, H2O, NH3. Декарбоксилазы.
5) Изомеразы: ферменты, катализирующие различные типы реакций изомеризации. Сюда относятся рацемазы и эпимеразы.
6) Лигазы (синтетазы) - ферменты, катализирующие синтез органических веществ из 2-х исходных молекул с использованием энергии АТФ. Название: «X-Y-лигаза». X и Y - исходные вещества. Например: глутомат-аммиак-лигаза.
Кроме всего этого все существующие ферменты (более 2000) имеют свой цифровой шифр, который присваивается по 4-х значному коду. Т. о. шифр каждого фермента состоит из 4-х цифр, разделенных точками и составляется по следующему принципу.
Первая цифра указывает на номер одногоиз классов ферментов.
Вторая цифра озночает подкласс, который характеризует тип связи, на которую действует фермент.
Третья цифра означает подподкласс, который характеризует химическую природу донора или акцептора, участвующего в реакции.
Четвертая цифра обозначает порядковый номер фермента.
Алкогольдегидрогеназа (АДГ); КФ: 1. 1. 1. 1.
Лактатдегидрогеназа (ЛДГ); КФ: 1. 1. 1. 27.
В основе классификации ферментов - тип катализируемой реакции
Большое число ферментов уже в начале XXвека поставило перед исследователями вопросы о номенклатуре и классификации ферментов. Отличительным признаком фермента в началеXXвека стало окончание «аза», которое использовали, добавляя его вначале к названию субстрата (amylum-крахмал - амилаза), а затем к названию реакции (дегидрирование - дегидрогеназы). Созданная Международным союзом химиков и биохимиков Комиссия по Ферментам (КФ) разработала основные принципы классификации и номенклатуры ферментов, которые были приняты в 1961 г. В основу классификации был положен тип катализируемой ферментом реакции. Все ферменты по этому признаку были разделены на 6 классов, в каждом из которых есть несколько подклассов.
1.Оксидоредуктазы - ферменты, которые катализируют реакции восстановления или окисления. Например алкогольдегидрогеназа, фермент, который окисляет этиловый спирт в уксусный альдегид. Второй фермент, известный как альдегиддегидрогеназа затем преобразовывает уксусный альдегид в ацетил КoA. Оксидоредуктазы часто требуют участия кофакторов, выполняющих роль промежуточных акцепторов водорода в приводимом ниже примере это НАД+.
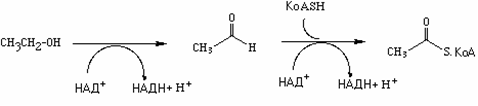
О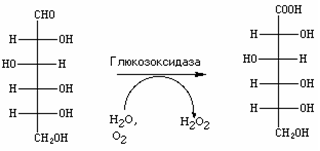 ксидазы
–разновидность оксидоредуктаз.
Так называются ферменты, использующие
кислород в качестве конечного акцептора
водородов. Примером может служить
глюкозоксидаза, которая окисляет
глюкозу в глюконовую кислоту.Промежуточным акцептором водородов
служит ФАД.
ксидазы
–разновидность оксидоредуктаз.
Так называются ферменты, использующие
кислород в качестве конечного акцептора
водородов. Примером может служить
глюкозоксидаза, которая окисляет
глюкозу в глюконовую кислоту.Промежуточным акцептором водородов
служит ФАД.
|
Оксидоредуктазы(1.0.0.0.) 1.1.0.0. Действуют на СН-ОН группы доноров 1.1.1.0. НАД+или НАДФ+в качестве акцепторов 1.1.1.1. Алкогольдегидрогеназа 1.14.0.0. Действуют на парные доноры при включении в один из них кислорода 1.14.15.0. Один из доноров восстановленный железо-серный белок и включение одного атома кислорода 1.14.15.1. Цитохром Р-450 1.14.15.5. Кортикостерон 18-монооксигеназа |
2.Трансферазы - ферменты, которые переносят функциональные группы от молекулы донора на молекулу акцептор. Примером могут служить метилтрансферазы, которые передает метиловую группу отS-аденозилметионина какому либо акцептору. Ниже показана реакция, катализируемая катехол-O-метилтрансферазой - ферментом, участвующим в метаболизме нейромедиаторов адреналина и норадреналина.

Еще один очень важный пример трансфераз – ферменты катализирующие перенос аминогруппы -трансаминазы.

Трансаминазы используют аминокислоту в качестве донора аминогруппы, которую они переносят на - кетокислоту, превращая соответственно аминокислоту – донор вкетокислоту и кетокислоту – акцептор в аминокислоту. Это используется для взаимопревращения некоторых аминокислот и позволяет аминокислотам вступать в пути метаболизма углеводов или липидов.
Трансферазами, которые будут часто упоминаться в биохимии, являются киназы, катализирующие перенос фосфата от макроэргической молекулы АТФ на субстрат. Существует множество киназ, играющих важную роль в метаболизме клеток.
|
Трансферазы(2.0.0.0.) 2.1.0.0.Переносят одноуглеродные группы 2.1.1.0. Метилтрансферазы 2.1.1.1. Никотинамид метилтрансфераза 2.1.1.45. Тимидилат синтаза 2.3.0.0. Ацилтрансферазы 2.3.1.6. холинацетил трансфераза |
3. Гидролазы-ферменты катализирующие биологические реакции гидролиза. Они разрывают ковалентные связи. присоединяя по месту разрыва элементы воды. Липазы, фосфатазы, ацетилхолинэстераза и протеазы - все это примеры гидролитических ферментов.
|
Гидролазы(3.0.0.0.) 3.1.0.0.Действуют на эфирные связи 3.1.1.0.Гидролазы эфиров карбоновых кислот 3.1.1.17. Ацетилхолинэстераза 3.2.1.0. Гликозидгидролазы 3.2.1.1. амилаза 3.2.1.2. -амилаза 3.4.0.0. Действуют на пептидные связи 3.4.21.0.Сериновые протеазы 3.4.21.1.Химотрипсин 3.4.21.4. Трипсин 3.4.21.5. Тромбин
|
4. Лиазы (десмолазы) –ферменты, которые катализируют распадC-C,C-OиC-Nсвязями негидролитическим путем с образованием двойных связей. Примером может быть фермент ДОФА декарбоксилаза, которая является ключевым ферментом в синтезе биогенных аминов адреналина и норадреналина.
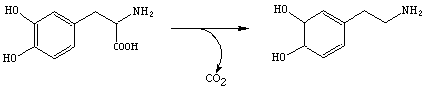
|
Лиазы(4.0.0.0) 4.1.0.0.Углерод-углерод лиазы 4.1.1.0.Карбокси лиазы 4.1.1.1. Пируватдекарбоксилаза 4.2.0.0. Углерод-кислород-лиазы 4.2.1.0. Гидролиазы 4.2.1.11. Енолаза 4.2.1.12. Фосфоглюконатдегидраза |
5. Изомеразы- ферменты, которые катализируют внутримолекулярные перегруппировки. При этом происходит взаимопревращение оптических геометрических и позиционных изомеров. Эпимеразы и рацемазы - примеры ферментов этого класса.
|
Изомеразы (5.0.0.0.) 5.1.0.0. Рацемазы и эпимеразы 5.1.1.0. Действуют на аминокислоты и их производные 5.1.1.1. Аланинрацемаза 5.3.0.0. Внутримолекулярные оксидоредуктазы. 5.3.1.0.Взаимопревращают альдозы и кетозы 5.3.1.9. Фосфоглюкоизомераза 5.3.1.20. Рибозоизомераза |
6. Лигазыкатализируют образованиеC-O,C-S,C-NилиC-Cсвязей, используя энергию гидролиза АТФ. Фосфат может или не может ковалентно связываться с продуктом реакции.
|
Лигазы (6.0.0.0) 6.1.0.0. Образуют С-О связи 6.1.1.0.Образуют молекулы аминоацил-тРНК и родственные им соединения. 6.1.1.1. Тирозил-тРНК синтаза 6.5.0.0. Образуют фосфоэфирные связи 6.5.1.1. ДНК-лигаза (АТФ -зависимая) 6.5.1.2. ДНК-лигаза (НАД+-зависимая) |
Комиссия по ферментам предложила и принципы номенклатуры ферментов. Рекомендуется использовать систематическую и рабочую номенклатуры. В основу систематической номенклатуры положен тот же принцип, что и для классификации – тип катализируемой реакции. На первый взгляд названия при этом становятся громоздкими, но зато из названия становится ясным, что делает фермент. Название состоит из двух частей: названия участников реакции ( в зависимости от класса это могут быть субстраты , промежуточные акцептоы) и типа катализируемой реакции с окончанием «аза».
Каждый фермент получает специфический кодовый номер-шифр фермента, отражающий его положение в классификации: первая цифра характеризует класс фермента, вторая –подкласс и третья подподкласс. Каждый подподкласс представляет собой список ферментов. Порядковый номер фермента в этом списке – четвертая цифра кода. На рис 1-1 показан шифр креатинфосфокиназы – КФ.2.7.3.2. Этот фермент катализирует реакцию фосфорилирования креатина. Систематическое название фермента АТФ: креатинфосфотрансфераза. Рабочее название этого фермента креатинкиназа или креатинфофокиназа
Ш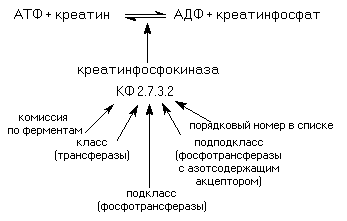 ифр
креатинфосфокиназы и место фермента
в классификации ферментов
ифр
креатинфосфокиназы и место фермента
в классификации ферментов
|
Заведующий кафедрой биологической химии, д.м.н., проф. |
Грицук А. И. |
___________ |
21.10.2006
Министерство здравоохранения Республики Беларусь
УО «Гомельский государственный медицинский университет»
Кафедра биологической химии
Обсуждено на заседании кафедры (МК или ЦУНМС)
Протокол № _________________200__года
ЛЕКЦИЯ по биологической химии
наименование дисциплины
для студентов _2__ курсалечебногофакультета
Тема Ферменты 5. Медицинская энзимология.
Время 90 мин.
Учебные и воспитательные цели:
Дать представление:
О медицинской энзимология: основные направления (энзимопатология, энзимодиагностика и энзимотерапия); применение ферментов в лабораторной диагностике; производственной практике и биотехнологии.
Об энзимопатиях: первичных и вторичных энзимопатии, степени клинического проявления; патогенезе энзимопатий – механизме развития вторичных метаболических блоков. Об энзимодиагностике: типы ферментов плазмы крови (клеточные, экскреторные, секреторные).
Об энзимотерапии: примеры; применении иммобилизованных ферментов, липосом, тенях эритроцитов, вирусных векторов.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф.Биологическая химия. М.: Медицина, 1990. С. 213–220; 1998. С. 345–353.
Николаев А. Я.Биологическая химия. М.: Высшая школа, 1989. С. 199–221.
Дополнительная
Марри Р. и др. Биохимия человека. М.: Мир, 1993. Т. 1. С. 111–126.
Филиппович Ю. Б. Основы биохимии. М.: Высшая школа, 1993. С. 403–415.
Ленинджер А.Основы биохимии. М.: Мир, 1985. Т. 2. С. 477–507.
МАТЕРИАЛЬНОЕ ОБЕСПЕЧЕНИЕ
1. Мультимедийная презентация.
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№ п/п |
Перечень учебных вопросов |
Количество выделяемого времени в минутах |
|
|
Медицинская энзимология. Основные направления (энзимопатология, энзимодиагностика и энзимотерапия). Применение ферментов в лабораторной диагностике, производственной практике и биотехнологии. |
30 |
|
|
Энзимопатии, классификации. Первичные и вторичные энзимопатии, степень клинического проявления. Патогенез энзимопатий – механизм развития вторичных метаболических блоков. Энзимодиагностика, цель, задачи. Типы ферментов плазмы крови (клеточные, экскреторные, секреторные). |
30 |
|
|
Энзимотерапия. Примеры. Иммобилизованные ферменты, липосомы, тени эритроцитов, вирусные векторы. Биотехнология. |
30 |
Всего 90 мин
Медицинская энзимология. Основные направления Применение ферментов в лабораторной диагностике, производственной практике и биотехнологии.
Медицинская энзимология - раздел клинической биохимии, который занимается изучением роли ферментов в заболеваниях, использование ферментов как лечебных препаратов и для диагностики.
Имеет 5 направлений:
1. Энзимопатология. (Изучение роли ферментов в развитие патологических процессов). Объект - изучения энзимопатиия.
2. Энзимодиагностика. (Изучение способов диагностики заболеваний путем определения активности ферментов).
3. Энзимотерапия. (Использование ферментов в качестве леарственных препаратов).
4. Инженерная энзимология. ( Использование ферментов в качестве технических и фармацевтических средств, в качестве реагентов).
5. Лабораторная диагностика. (Выделение ферментов в малых количествах).
Энзимопатии. Патогенез энзимопатий. Энзимодиагностика, цель, задачи. Типы ферментов плазмы крови.
Энзимопатия - заболевания, в основе которого лежат генетические и др. изменения ативности ферментов.
Причины:
По классификации академика Покровского энзимопатиии делятся на:
1) наследственные (генетически детерминированы - точечные мутации, хромосомные аберрации ---> серповидноклеточная анемия, фенилкетонурия, галактоземия, альбинизм);
2) алиментарные - связаны с пищевыми факторами: дефицит белка, микроэлементов, гипо- и авитоминозы, несбалансированное питание. Употребление недоброкачественной пищи (красители, консерванты);
3) токсические - связаны с ингибированием ферментов пестицидами, гербицидами, лекарствами, выбросами машин, заводов (например тетрациклин блокирует рибосомальный цикл гепатоцитов в печени).
По современной классификации:
1) первичные (врожденные, наследственные);
2) вторичные (приобретенные: алиментарные и токсические).
Причины первичных энзимопатий:
1. Точечные мутации гена, кодируюшего фермент.
2. Наличие ингибитора или отсутствие активатора при синтезе фермента.
3. Генетические дефекты синтеза кофермента.
4. Нарушение процессинга белка.
5. Патология или отсутствие матрицы ДНК и РНК.
Причины вторичных энзимопатий:
1. Нарушения энергообеспечения (уменьшение АТФ, ГТФ - нарушение синтеза апофермента).
2. Недостаток аминокислот (белковое голодание).
3. Отсутствие или недостаток кофермента: витаминов, микроэлементов, нарушение ресорбции витаминов в ЖКТ.
4. Все причины гиповитаминозов.
5. Клеточная деструкция разного генеза.
Все инфекционные болезни (вирусные, бактериальные и паразитарные) протекают с расстройством ферментных систем, это связано с выделением экзо- и эндотоксинов, блокирующих ряд ферментов.
Другой причиной является гипо- и гиперфункция эндокринных желез.
Также причиной может быть резкое изменение условий среды, в которой работает фермент. (Ацидоз или алколоз).
Примеры энзимопатий:
В условиях любого ферментативного блока, активируются минорные пути метаболизма:
схема
ФПК (фенилпировиноградная кислота) - является конкурентным ингибитором в ткани мозга и блокирует аэробные энергодающие пути мозга, вызывающие дефицит энергии. Накапливается в мозге и вызывает дегенерацию, сопровождается расстройством психической деятельности.
Алкаптонурия - болезнь «черных пеленок», моча на воздухе чернеет за счет окисления неразложившейся гомогентизиновой кислоты.
ТИР ----> ДОФАмин ----> катехоламины схема
отсутствие ----> меланина в коже, волосах и сетчатке.
Синдром «кленового сиропа» (моча имеет сладковатый привкус): возникает вследствие дефекта фермента метаболизма а/к с разветвленными радикалами (вал, лей, изолей), концентрация этих а/к увеличивается, активируются минорные метаболические пути и образуется альфа-кетокислоты, аналоги этих а/к.
Гиперуринемия (болезнь Леша-Нихана) - повышенное содержание мочевой кислоты в крови, вызванное недостаточностью гуанин-гипоксантин фосфорибозинтрансферазы.
Доброкачественная желтуха новорожденных связана с понижением активности глюкуронилтрансферазы.
Злокачественная желтуха - дефект глюкоронилтрансферазы.
Гемофилия А (дефицит VIII фактора свертываемости крови).
-//- В (дефицит IX фактора).
-//- С (дефицит XI фактора).
В основе всех ферментопатий лежит увеличение концентрации S для аномального фермента, активация минорных путей метаболизма, приводящих к образованию токсических веществ, вызывающих вторичные патологические блоки.
E
S - X --> P
А дефицит P выражается снижением интенсивности последующих реакций.
Степень проявления энзимопатий.
1. Бессимптомные, не имеющие никаких проявлений (фруктозурия).
2. Слабо выраженные (проявления средней степени: легкие формы сахарного диабета; генетические дефекты бета-структуры Hb, гипоксия).
3. Ярко-выраженные (несовместимые с жизнью) - заболевание проявляется с первых дней жизни: болезнь «кленового сиропа».
Энзимодиагностика.
Еще Вольгемут показал, что при заболевании поджелудочной железы в моче и крови обнаруживается высокая активность амилазы.
Так при заболевании костей имеет место высокая активность щелочной фосфатазы, при заболевании простаты - кислой фосфатазы.
Таким образом энзимодиагностика базируется на идее органоспецифичности и компартментолизации ферментов в клетке.
При заболевании увеличивается проницаемость мембран и вследствие нарушения градиента концентраций ферментов между внутриклеточной и межклеточной средами, ферменты выходят из клетки и попадают в кровь, мочу.
Наилучшим источником диагностических ферментов является сыворатка крови. Активность ферментв в сыворотке прямо пропорциональна поражению органа: чем больше активность - тем серьезней повреждение. Нужно отметить, что все антикоагулянты являются ингибиторами плазменных ферментов.
Энзимодиагностика имеет 2 направления:
1. Ранняя диагностика.
Так при гепатитах активность трансаминаз (АсАТ АлАТ) повышается гораздо раньше, чем билирубин проникает в ткани и вызовет желтуху, и значительно раньше, чем возникает недомагание.
Путем определения активности АсАТ и АлАТ можно диагнозировать гепатит за 2-е недели до появления желтухи.
2. Дифференциальная дигностика.
Так например заболевание печени делются на 3 группы.
Кроме этого нужно отметить, что АсАТ АлАТ одинаково представлены в печени и миокарде, поэтому при повреждении того и другого активность их увеличивается. Но при заболевании сердца (инфаркте) преобладает активность АсАТ. При отсром гепатите - АлАТ.
В клинике для диф. диагностики ифаркта миокарда используется коэф. Де Ритиса АсАТ
Коэф. Де Ритиса = ---- = 1,5 - 2
АлАТ
При гепатите коэф. < 0,6 (увеличивается активность АлАТ).
Если коэф. > 2 (увеличивается активность АсАТ), то это инфаркт миокарда; уровень держится 2-3 суток, к концу 1 недели падает.
Для оценки степени тяжести заболеваний также определяется активность ферментов. Так при мягких формах гепатита сначала увеличивается ативность фермента цитоплазмы гепатоцита - дегидрогеназы. При тяжелых формах - увеличивается активность митохондриального фермента глутаматдегидрогеназы.
Этим же путем можно дифференцировать гепатит и некроз печени. При гепатите наблюдается высокая активность билирубина и АлАТ, при некрозе - активность билирубина увеличивается, а АлАТ уменьшается.
Следует отметить, что при заболеваниях может наблюдаться 3 состояния ферментов:
1) гипоферментемия (снижена активность в плазме). Наблюдается при поражении того органа в котором синтезируется данный фермент, например при гепатите уменьшается активность холинэстеразы, синтезируемой в гепатоцитах. Так, на высоте заболевания панкреотитом, снижается активность амилазы, что ведет к некрозу.
2) Нормоферментемия, может сопровождаться дисферментемией, характерно для текущих процессов - цирроз печени.
3) Гипеферментемия встречается чаще всего.
Некоторые примеры использования измерения активности ферментов в диагностике
Фосфатаза - фермент, который не имеет выраженной субстратной специфичности к органическим эфирам фосфорной кислоты. По рН-оптимумам различают кислую и щелочную фосфатазы. Высокой активностью кислой фосфатазы отличается предстательная железа. При раке предстательной железы активность этого фермента увеличивается в сыворотке крови. Щелочная фосфатаза находится во многих органах, особенно в костной ткани и печени. Подобно ЛДГ существуют органоспецифические изоферменты щелочной фосфатазы. Печеночная фосфатаза, хотя и является клеточным ферментом (лизосомы), в небольших количествах выделяется с желчью в кишечник. Поэтому при нарушении оттока желчи (желчекаменная болезнь, опухоль) часть фосфатазы всасывается в кровь. Этот эффект может иметь место при повреждении печеночных клеток. Повреждение печени нередко сопровождаются желтухами. Энзимодиагностическое исследование кислой фосфатазы используется для дифференциальной диагностики желтух. Такая диагностика важна для врача при выборе методов лечения. Дело в том, что разные типы желтух требуют принципиально разного лечения: хирургическое - в случае закупорки желчного протока и консервативное - при паренхиматозном повреждении печени. Активность щелочной фосфатазы повышается при патологических процессах в костной ткани (метастазы в кость или распад костей). Различить изоферменты фосфатазы из печени и костей можно на основании их различной чувствительности к мочевине. После добавления определенного количества мочевины остаточная активность связана с печеночной фосфатазой, ибо она не ингибируется мочевиной.
|
Таблица 2-4 Примеры ферментов, используемых для энзимодиагностики заболеваний.. | ||
|
Фермент |
Основные источники |
основное клиническое приложение |
|
Кислая фосфатаза |
Предстательная железа, эритроциты |
Рак предстательной железы |
|
Аланин аминотрансфераза |
Печень, скелетная мышца, сердце |
Болезни печеночной парехимы |
|
Альдолаза |
Скелетная мышца, сердце |
Болезни мышц |
|
Щелочная фосфатаза |
Печень, кость, слизистая оболочка кишечника, плацента, почки |
Болезни костной ткани, болезни печени |
|
Амилаза |
Слюнные железы, поджелудочная железа, яичники |
Заболевания поджелудочной железы |
|
Аспартат аминотрансфераза |
Печень, скелетная мышца, сердце, почки, эритроциты |
Инфаркт миокарда, болезни печеночной паренхимы, мышц |
|
Холинэстераза |
Печень |
Отравление фосфорорганическими инсектицидами, болезни печени |
|
Креатинкиназа |
Скелетная и гладкая мышцы, мозг, сердце, |
Инфаркт миокарда, болезни мышц |
|
Глутаматдегидрогеназа |
Печень |
Болезни печеночной паренхимы |
|
-Глутамил транспептидаза. |
Печень, почки |
Гепатобилиарные болезни, алкоголизм |
|
Лактатдегидрогеназа |
Сердце, печень, скелетная мышца, эритроциты, тромбоциты, лимфатические узлы |
Инфаркт миокарда, гемолиз, болезни печеночной паренхимы |
|
5 ' нуклеозидаза. |
Гепатобилиарный тракт |
Гепатобилиарные болезни |
|
Сорбитдегидрогеназа . |
Печень |
Болезни паренхимы печени |
|
Трипсин (оген) |
Поджелудочная железа |
Заболевания поджелудочной железы |
Глутамилтранспептидаза (ГТ) является печеночным ферментом и имеет значение для диагностики нарушений печени. При паренхиматозных заболеваниях печени, расстройстве функции желчевыделительной системы часто намного раньше других клеточных ферментов в сыворотке крови повышается активностьглутамилтранспептидазы (ранняя диагностика). Она же остается повышенной гораздо дольше всех остальных ферментов.
Энзимотерапия. Примеры. Иммобилизованные ферменты, липосомы, тени эритроцитов, вирусные векторы. Биотехнология.
Ферменты, которые обнаруживаются в норме в плазме крови делятся на 3 группы:
1. Секреторные - они синтезируются в печени и выделяются в плазму крови: ферменты гемостаза, холинэстераза.
2. Индикаторные (клеточные). Одни из них локализованы в цитоплазме (ЛДГ), другие - в митохондриях (ГДГ), третьи - в лизосомах (кислая фосфотаза). Большая часть индикаторных ферментов в плазме определяется лишь в следовых количествах, и только при поражении каких-либо тканей их активность резко возрастает.
3. Экскреторные (ИАП, щелочная фосфотаза) - синтезируются в основном в печени и выделяются с желчью. При патологических процессах их выделение с желчью нарушается и активность в плазме возрастает.
Принципы диагностики энзимопатий.
1. Определение концентраций в биологических жидкостях, тканях субстратов и продуктов тех ферментов, активность которых снижена.
2. Определение активности фермента, который вызывает энзимопатию.
3. Определение концентраций метаболитов минорных путей метаболизма: ФПК, ФУК.
4. Клиническая диагностика: симптомы, заметные глазом - глаза, внешний вид.
Энзимотерапия.
Способ лечения с помощью ферментов - энзимотерапия.
Применяетя заместительная терапия при недостаточности ферментов ЖКТ (желудочный сок, продукты поджелудочной железы, стимулирующие HCl, алахол - стимулятор липазной активности.
Ферменты применяются для апликаций, ингаляций при гнойных заболеваниях легких.
Ферменты: РНКазы, ДНКазы, гиалуронидаза, коллагеназы, эластазы используются для обработки ран, воспалительных очагов, ожогов, для устранения отеков.
Для лечения заболеваний ССС применяются кашикрины (для снижения кровяного давления), стрептодеказа (ведет к снижению зоны инфаркта миокарда).
В последнее время применяются иммобилизованные ферменты, т. е. фиксированные на чем либо. Такие ферменты обладают повышенной стабильностью, сниженной антигенностью и более длительным действием в организме.
В настоящее время открыт новый способ введения иммобилизованных ферментов, при помощи микосом. Микосомы - мелкие частицы эмульгированного жира содержащие ферменты, окруженные несколькими фосфониаледными оболочками.
Липосомы - хорошие носители лекарств, они биосовместимы, не вызывают иммунологических реакций и с их помощью можно вводить ферменты внутрь клеток. С помощью микосом были введены, растворяющие мельчайшие шарики, необходимые для трансферации эндотелия в месте образования тромба.
Предпринимались попытки применения ферментов для лечения злокачественных опухолей, например аспарагиназы при лечении лимфогранулематоза. Этот фермент разрушает АСН, является незаменимым фактором для лейкозных клеток.
|
Заведующий кафедрой биологической химии, д.м.н., проф. |
Грицук А. И. |
___________ |
21.10.2006
Министерство здравоохранения Республики Беларусь
УО «Гомельский государственный медицинский университет»
Кафедра биологической химии
Обсуждено на заседании кафедры (МК или ЦУНМС)
Протокол № _________________200__года
ЛЕКЦИЯ по биологической химии
наименование дисциплины
для студентов _2__ курсалечебногофакультета
Тема Биологическое окисление 1. Цикл Кребса.
Время 90 мин.
Учебные и воспитательные цели:
Дать представление:
Об истории развития учения о биологическом окислении (БО) и роли отечественных и зарубежных ученых в развитии учения о БО.
О современном представления о БО: окислительно-восстановительный процесс, окислительно-восстановительная реакция, окислительно-восстановительный потенциал (ОВП). Об основных этапах БО. О принципах БО. Об организме как антиэнтропийной системе. О строении АТФ, природе макроэргичности. АТФ-азном цикле.
О митохондрии (МX): строении, функциях, сравнительной характеристике мембран митохондрий; о характеристике ферментов мембран, межмембранного пространства, МX матрикса.
О цикле трикарбоновых кислот (ЦТК), истории открытия, реакциях, ферментах, коферментах, субстратах. О биологической роли ЦТК (энергетической, пластической, регуляторной). О регуляции ЦТК (субстратами, отношением NAD/NADH и ADP/ATP, O2, ионамиCa,pH). О метаболонах ЦТК.
ЛИТЕРАТУРА
Березов Т. Т., Коровкин Б. Ф.Биологическая химия. М.: Медицина, 1990. С. 213–220; 1998. С. 305–317.
Николаев А. Я.Биологическая химия. М.: Высшая школа, 1989. С. 199–221.
Дополнительная
Филиппович Ю. Б. Основы биохимии. М.: Высшая школа, 1993. С. 403–438.
Марри Р. и др. Биохимия человека. М.: Мир, 1993. Т. 1. С. 111–139.
Ленинджер А.Основы биохимии. М.: Мир, 1985. Т. 2. С. 403–438, 508–550.
Албертс Б. и др., Молекулярная биология клетки. М.: Мир, 1994. Т. 1. С. 430–459.
Скулачев В.П. Энергетика биологических мембран. М.: Наука. 1989.
МАТЕРИАЛЬНОЕ ОБЕСПЕЧЕНИЕ
1. Мультимедийная презентация.
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№ п/п |
Перечень учебных вопросов |
Количество выделяемого времени в минутах |
|
|
История развития учения о биологическом окислении (БО). Роль М. В. Ломоносова, А. Лавуазье, Ф. Шейнбайна, А. Н. Баха, В. И. Палладина, В. А. Энгельгардта, А. Сент-Дьерди и др. в развитии учения о БО. |
15 |
|
|
Современные представления о БО: окислительно-восстановительный процесс, окислительно-восстановительная реакция, окислительно-восстановительный потенциал (ОВП). Понятие об окислении и восстановлении. Основные этапы БО и их энергетическая ценность. Принципы БО. Организм как антиэнтропийная система. Строение АТФ, природа макроэргичности. АТФ-азный цикл. |
25 |
|
|
Митохондрия. Строение, функции, сравнительная характеристика мембран митохондрий. Характеристика ферментов мембран, межмембранного пространства, МX матрикса. |
20 |
|
|
ЦТК, история открытия, реакции, ферменты, коферменты, субстраты. Биологическая роль ЦТК (энергетическая, пластическая, регуляторная). Регуляция ЦТК (субстратами, отношением NAD/NADH и ADP/ATP, O2, ионамиCa,pH). Метаболоны ЦТК. |
30 |
Всего 90 мин
История развития учения о биологическом окислении
Еще древние философы отмечали взаимосвязь между процессами жизнедеятельности и дыханием. Они также провели параллель между дыханием и горением.
Платон утверждал, что воздух нужен для охлаждения внутреннего жара сгорающего вещества.
Аристотель утверждал, что воздух нужен для поддержания внутреннего горения.
В XVII - XVIII вв широкое признание получила теория горючего начала - флогистона, сформулированная Штаммом. Эта теория объясняла процессы горения выделениями их горящего тела особого невесомого в-ва, и была опровергнута Ломоносовым и Лавуазье, которые открыли закон сохранения энергии.
В XVIII веке с развитием физики газов, с появлением соответствующей аппарутуры стали проводить опыты по сжиганию веществ в замкнутом пространстве. В это время Шталем была сформулирована теория флагистона (горючего начала), согласно которой все вещества, подвергающиеся окислению состоят из оксида и флагистона.
В середине XVIII века было установлено:
1) процесс горения идет в воздушной среде с высокой температурой, дыхание - в среде с пониженной температурой;
2) при дыхании, как и при горении выделяется тепло, но в незначительных количествах;
3) конечные продукты CO2и H2O.
В 1751 году Ломоносов подробно рассмотрел процессы горения и окисления.
В 1774 году Лавуазье повторил опыты Ломоосова и показал, что процессы горения и дыхания идентичны, т. к. образуются идентичные продукты.
Лавуазье назвал дыхание медленным горением и показал процесс сгорания Гл в организме:
C6H12O6 + 6O2 ------> 6CO2 + 6H2O + Q
В начале XIX века стали известными катализаторы, с помощью которых осуществлялись процессы окисления. Это были металлы, обладающие «внутренней силой».
В середине XIX века немецкий ученый Шейнбайн, открывший озон, предположил, что в организме образуется озон и он используется в реакциях окисления.
После работ Лавуазье в науке господствовало мнение о тождестве горения и медленного окисления питательных вкществ в организме. Вместе с тем было ясно, что БО протекает в необычных условиях:
- при пониженной температуре;
- без пламени;
- и в присутсвии большого количества H2O (75% - 80% ткани).
В XIX веке появилось понятие о ферментах и причину своеобразного течения БО попытались объяснить «активацией» кислорода в клетках организма.
Одна из теорий была выдвинута Бахом, который считал, что «активация» молекулярного кислорода происходит в результате разрыва связи и присоединения к ферментам оксигеназам (А), способным к аутооксидации:
O A + O2 -----> A |
O перекись
O
A | + SH2 -----> S + A + H2O2 O субстарат
3 положения Баха:
1. Наличие высокоактивной оксидазы, но это не было обнаружено.
2.В тканях должна быть высокая концентрация H2O2, но это тоже не было обнаружено.
3. Высокая активность ферментов, разлагающих H2O2; это было обнаружено, существует 2 фермента:
каталаза
2H2O2 ------------> 2H2O + O2
Существует и другой механизм разложения H2O2:
2GSH + H2O2-----> 2H2O (пероксидаза) или глутатион |
SH2 + H2O2 -----> S + 2H2O ---
Эта теория да и все остальные основывались на неправильном представлении об ОВР. Окислительный процесс рассматривался как процесс взаимодействия любого вещества с кислородом. То есть кислород - это окислитеоь.
К концу XIX века с разхвитием физики ядра и накопления знаний о структуре вещества, было установлено, что не все процессы окисления требуют для своей реализации наличие кислорода.
Кроме этого теория Баха основывалась на том, что в организме имеется большое количество ароматических соединений, на самом же деле их очень мало, в оновном глюкоза.
Согласно современных представлений ОВР - это процесс перемещения электронов и протонов от донора (восстановителя) - это процесс окисления - к акцептору (окислителю) - процесс восстановления.
Количественной мерой ОВР является величина ОВП. В начале точки отсчета стандартного потенциала взят ОВП водорода.
В 1912 году была сформулирована теория Палладина-Виланда, согласно которой в организме есть промежуточные вещества, способные акцептировать электроны и протоны от субстрата с последующей передачей электронов и протонов на кислород, по этой теории весь процесс БО можно разбить на 2 этапа:
1) анаэробный - передача электронов и протонов с субстрата на промежуточное вещество;
2) аэробный - передача электронов и протонов с промежуточного вещества на кислород.
Палладин предпологал, что существует несколько промежуточных переносчиков, позволяющих организму поэтапно освобождать химическую энергию и кислород выступает в качестве конечного акцептора электронов и протонов.
1 анаэробный этап:
SH2+ ----> S +
2 аэробный этап:
½ O2 -----> + H2O
Роль промежуточных переносчиков (хромогенов) выполняют коферменты (НАД; НАДФ; ФАД; ФМН) оксидоредуктаз.
В последствии развития учения о БО, шло по пути развития знаний о хромогенах.
В 1925 году были открыты гистогематины (цитохромы).
В 1932 году академик Энгельгардт показал, что процесс окисления идет с образованием АТФ (окислительное фосфорилирование).
В 1945 году Ленинджер и Кенеди впервые показали, что процесс окисления веществ, цикл Кребса локализованы в митохондриях.
Современные представления о БО базируются на сущности трактовки ОВП, а также БО основано на общих законах термодинамики:
1 закон - закон сохранения энергии: энергия никуда не исчезает, а только переходит из одной формы в другую, т. е. сохраняется.
2 закон - все тела и химические процессы стремятся к минимуму энергии, к состоянию покоя и беспорядка, т. е. к энтропии.
С термодинамической точки зрения - организм человека - антиэнтропийная машина, открытая система, которая обменивается с окружающей средой веществом и энергией. Основа ее жизнедеятельности - обмен веществ (метаболизм).
Современные представления о БО. Основные этапы БО. Строение АТФ, природа макроэргичности.
Обмен веществ и энергии - закономерный порядок превращения вещества и энергии в живых организмах, направленный на их сохранение и самовоспроизведение. Обмен веществ и обмен энергии тесно связаны и представляют собой диалектическое единство.
Вся совокупность химических реакций, протекающих в живых органихмах, включая усвояемость веществ, поступающих извне (ассимиляция) и их расщепление (диссимиляция) вплоть до образования конечных продуктов, подлежащих выделению, составляет сущность и содержание обмена веществ.
Ассимиляция - одна из сторон обмена веществ.
Ассимиляция включает огромное количество химических превращений, приводящих к использованию органических и неорганических веществ, поступающих из внешней среды для построения специфических для данного организма белков, НК, липидов, углеводов. Процесс ассимиляции обеспечивает рост, развитие, обновление организма и накопление запасов, используемых в качестве источника энергии.
Диссимиляция - противоположная ассимиляции сторона обмена веществ: разрушение органических соединений с превращением их в простые вещества (в основном H2O, CO2, NH3).
Промежуточный обмен - превращение веществ в организме с момента поступления их в клетки до образования конечных продуктов.
Попав внутрь клетки, питательное вещество метаболизируется - претерпевают ряд химических изменений, катализируемых ферментами. Определенная последовательность таких химических изменений называется метаболическим путем, а образующиеся прмежуточные продукты - метаболитами.
Различают 2 стороны промежуточного обмена - анаболизм и катоболизм. Анаболические реакции направлены на образование и обновление структурных элементов клеток и тканей. Эти реакции преимущественно восстановительные, соправаждаются затратой свободной энергии.
Катоболические превращения - процессы расщепления сложных молекул, как поступивших с пищей, так и входящих в состав клетки до простых компонентов. Эти реакции обычно окислительные, сопроваждаются выделением свободной энергии.
Обе стороны промежуточного обмена тесно взаимосвязаны во времени и пространстве.
Боилогическое окисление - это совокупность биохимических реакций, приводящих к образованию полезной энергии за счет деградации компонентов пищи.
Принципиальной особенностью БО или тканевого дыхания является то, что оно протекает постепенно, через многочисленные промежуточные стадии, т. е. происходит многократная передача протонов и электронов от донора к акцептору.
Субстраты биологического окисления.
Субстратом БО является любое вещество, способное поставлять электроны и протоны, энергия которых трансформируется в полезную конвертируемую форму.
Субстраты БО: метаболиты восстанавливающие НАД, ФАД, служащие предшественниками субстратов, зависящие от дегидрогеназ Гл, АК.
Схема энергетического обмена.
Основные компоненты пищи - белки, липиды и углеводы проходят 3 этапа энергетического обмена:
1. ЖКТ - происходит деполимеризация сложных соединений: крахмал и гликоген -----> Гл
дисахариды и олиго -----> до моносахаридов
белки -----> до пептидов и АК.
2. С момента поступления мономеров в клетку начинается цитозольный этап: происходит дальнейший распад мономеров и унификация субстратов, превращение их в общие соединения:
Углеводы все идут в Гл.
Липиды ----> Гн и ЖК.
3. Митохондриальный - унификация субстратов продолжается в митохондриальном матриксе, тут субстраты подвергаются окислению путем вовлечения в цикл Кребса, который снимает с них электроны и протоны и трансформирует их энергию в конвертируемую форму АТФ.
Схема образования субстратов биологического окисления.
БЕЛКИ УГЛЕВОДЫ ЛИПИДЫ Энергия
| | | | |
АК Гл Гн ЖК |1; 0,5%
|
|
ЩУК |
| 3ФГА | | ПВК --- | | АцКоА |
> |
лактат |
| | | |2; 2,5% | | | | |
|
|
Цикл Кребса NAD 1/2O2 -------- АДФ + Фн |
| Цитрат NAD.H2 -> H2O АТФ работа |
|
----------------- |
| | | | | 3; 97% | | | ---------- |
Природа макроэргичности АТФ.
Роль АТФ - хранилище биологической энергии. В 1 молекуле АТФ имеется 2 макроэргические связи. При их расщеплении высвобождается 32 кДж энергии.

АТФ присутствует в клетках в диссоциированной форме: АТФ4-АТФ4- ------> АДФ3-+ Фн2-+ Н+
10-310-310-310-7
Т. о. всякая работа в клетке сопровождается образованием H+, которые захватываются буферами.
1 причина макроэргичности: т. к. концентрация АТФ, АДФ и Фн одинакова (по 10-3 моль), а концентрация Н+ = 10-7 моль,
согласно закону соотношения действующих масс равновесие сдвинуто вправо.
2 причина: в структуре АТФ имеется 3 фосфата и 2 ангидридные связи, за счет этого на хвосте молекулы АТФ создается конфармационная напряженность, возникает сила электростатического отталкивания и АТФ отдает молекулу фосфата. И при этом она переходит в более выгодное состояние АДФ + Фн, которое более устойчиво, это 3-я причина макроэргичности.
В клетках АТФ присутствует в виде магниевой соли. Существует точка зрения, что уровень Mg2+ отражает уровень АТФ.
В 1940-41 гг немецким биохимиком Фрицем Липманом была создана концепция АТФ-азного цикла: в процессе фото- или хемосинтеза энергия депонируется в форме АТФ. Синтез АТФ в организме происходит из АМФ:
Фн
АМФ + Фн ------> АДФ -------> АТФ
- HOH
фото -> АТФ ----------> осмотическая работа
синтез | ----> транспорт
хемо ---АДФ + Фн<-----> электрическая работа
----> химическая работа
----> тепловая работа
----> механическая и световая работа.
«Энергетической валютой» клетки является АТФ
Такое «центральное» расположение молекулы АТФ позволяет ей выполнять роль донора высокоэнергетического фосфата для соединений, расположенных ниже в таблице, превращаясь при этом в АДФ, а АДФ - роль акцептора высокоэнергетического фосфата у соединений, расположенных выше. Цикл АТФ/АДФ связывает, тем самым, процессы генерирующие «~Р» с процессами, использующими «~Р». Сумму всех адениловых нуклеотидов в клетке (АТФ,АДФ и АМФ) называют адениловой системой. Процессы гидролиза и синтеза АТФ происходят с высокой скоростью, поскольку общий фонд АТФ очень маленький и для поддержания процессов жизнедеятельности в клетке его хватает только на несколько секунд.
В клетках организмов животных есть три основных источника ~Pдля синтеза АТФ.
окислительное фосфорилирование – механизм образования АТФ, использующий для этого энергию градиента электрохимического потенциала, возникающего на внутренней мембране митохондрий.
Субстратное фосфорилирование – механизм синтеза АТФ, использующий энергию макроэргических соединений, образующихся в процессе метаболизма (1,3- дифосфоглицериновая кислота, сукцинил-КоА и т.д.).
Синтез АТФ с использованием макроэргов, выполняющих своеобразную роль молекул – депо макроэргических связей (креатинфосфат).
Тому, каким образом живые системы преобразуют энергию поступающих из внешней среды химических соединений, в энергию макроэргических соединений и посвящена значительная часть курса биохимии.
Митохондрия. Строение, функции, сравнительная характеристика мембран митохондрий. Характеристика ферментов мембран, межмембранного пространства, МX матрикса.
Митохондрии постоянные органеллы всех клеток (кроме эритроцитов) имеют 2 мембраны:
|
Признак |
Внутренняя мембрана |
Наружная мембрана |
|
1. Форма |
Складчатая (кристы) |
Гладкая |
|
2. Плотность |
1,2 |
1,1 |
|
3. ФЛ/Белки |
0,27/0,73 |
0,82/18 |
|
4. Проницаемость |
Высокоселективная |
Низкоселективная (даже большие молекулы проникают свободно) |
|
Содержание: | ||
|
5. Кардиолипина |
Высокое |
Низкое |
|
6. Фосфоинозитола |
Низкое |
Высокое |
|
7. Холестерина |
Низкое |
Высокое |
|
8. Ферменты |
СДГ и компоненты ДЦ |
МАО, ферменты синтеза ЖК |
Межмембранное пространство: в нем активны аденилаткиназа и нуклеозиддифосфаткиназа.
В процессах старения генома митохондрии мигрируют в ядро, т. е. возникают летальные мутации, связанные с деформацией митохондриальных белков генерирующих АТФ.
ЦТК, история открытия, реакции, ферменты, коферменты, субстраты. Биологическая роль, регуляция ЦТК. Метаболоны ЦТК.
Цикл трикарбоновых кислот или цикл лимонной кислоты был открыт Гансом Кребсом в 1937 г. Он брал измельченные мышцы голубя, добавлял на них трикарбоновые кислоты и определял скорость дыхания, те трикарбоновые кислоты, которые составляют цикл Кребса усиливают дыхание.
Цикл Кребса - исходный субстрат ацетил КоА, который взаимодействует с ЩУК под действием фермента цитратсинтетазы.
За один оборот цикла Кребса происходит полное окисление одной молекулы ацетил-КоА. Для непрерывной работы цикла необходимо постоянное поступление ацетил-КоА, а коферменты НАД и ФАД должны снова окисляться. Это и происходит в ДЦ.
Освобождающаяся при окислении ацетил-КоА энергия, расходуется на образование макроэргических связей АТФ.
Из 4 пар атомов водорода, 3 пары переносятся через НАД и одна пара через ФАД. На каждую пару атомов водорода в системе БО образуется 3АТФ (1НАДН2 = 1АТФ). Следовательно, всего 9АТФ; одна пара атомов попадает в систему БО через ФАД, - в результате образуется 2АТФ. Кроме этого в ходе сукцинаткиокиназной реакции образуется 1ГТФ = 1АТФ. Поэтому в целом, в ходе цикла Кребса образуется 12АТФ.
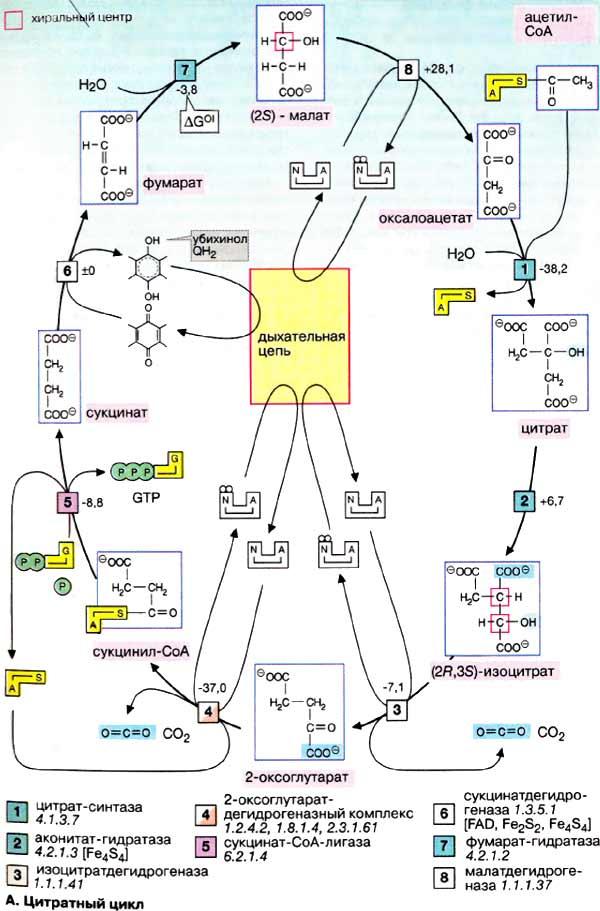
Биологическое значение ЦТК.
ЦТК - универсальный компонент БО, который образуется на принципе унификации, что имеет огромное значение, потому что организм не может точно дозировать потребность в каждом субстрате. Унификация позволяет уравновешивать и оптимизировать соотношение основных субстратов, т. е. если имеется избыток углеводов, то часть их перекачивается в липиды, если избыток белка, то тоже - в липиды и углеводы.