

40 Вопрос
Уравнение Ван-дер-Ваальса
ВАН-ДЕР-ВААЛЬСА УРАВНЕ́НИЕ, уравнение состояния, описывающее свойства реального газа. Предложено Й. Д. Ван-дер-Ваальсом в 1873 г. Широко используется для качественного анализа поведения реальных газов и жидкостей. В модели реального газа Ван-дер-Ваальса молекулы рассматриваются как абсолютно твердые слабо притягивающиеся упругие сферы определенного диаметра.
Уравнение Ван-дер-Ваальса количественно определяет свойства реальных газов лишь в небольшом интервале температур и давлений: в области относительно высоких температур и низких давлений, так как входящие в него экспериментально определяемые константы являются функциями температуры.
Для моля газа объемом V при температуре Т и давлении р, уравнение Ван-дер-Ваальса имеет вид:
(p+a/Vm 2)(Vm - b) = RT,
где: R — газовая постоянная,
a и b — экспериментальные константы, учитывающие отклонение свойств реального газа от свойств идеального газа.
Член a/V2 имеет размерность давления и учитывает притяжение между молекулами газа за счет ван-дер-ваальсовых сил. Действие сил притяжения газа приводит к появлению дополнительного давления на газ, называемого внутренним давлением. По вычислениям Ван-дер-Ваальса, внутреннее давление обратно пропорционально квадрату молярного объема, т. е. рвн = a/Vm2, где а — постоянная Ван-дер-Ваальса, характеризующая силы межмолекулярного притяжения, Vm — молярный объем.
Константа b является поправкой на собственный объем молекул газа и учитывает отталкивание молекул на близких расстояниях. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объем других молекул, приводит к тому, что фактически свободный объем, в котором будут двигаться молекулы реального газа, будет не Vm, а Vm-b, где b — объем, занимаемый самими молекулами. Объем b равен учетверенному собственному объему молекул.
Константы а и b обычно определяются из экспериментальных данных, и эти величины постоянны для каждого газа. Для их определения записывают уравнения для двух известных из опыта состояний газа и решают эти уравнения относительно а и b. При больших объемах V можно пренебречь обеими поправками и уравнение Ван-дер-Ваальса переходит в уравнение состояния идеального газа (см. Клапейрона уравнение).
Несмотря на то, что уравнение Ван-дер-Ваальса является приближенным и количественно описывает свойства реальных газов лишь в области высоких температур и низких давлений, качественно оно позволяет описывать поведение газа и при высоких давлениях, конденсацию газа в жидкость. Уравнение Ван-дер-Ваальса также описывает критическое и метастабильное состояние системы жидкость-пар.
41 Вопрос
Сравнение изотерм Ван-дер-Ваальса с экспериментальными. Критическое состояние.
Изотермы Ван-дер - Ваальса на pF - диаграмме с повышением температуры поднимаются вверх. Это сопровождается сближением объемов YI и F2 определяющих соответственно объемы жидкой и газообразной фазы. Совпадение жидкой и газообразной фазы произойдет в точке А ( см. рис. 10), которая называется критической. Эта точка определяется точкой перегиба изотермы, температура которой называется критической.
Изотерма Ван-дер - Ваальса, соответствующая критической температуре Ткр вещества, имеет точку перегиба - критическую точку.
Сравнение изотерм Ван-дер - Ваальса ( см. рис. 13.7) с эксперя-ментальными изотермами реальных веществ ( например, с рис. 13) показывает, что изотермы Ван-дер - Ваальса охватывают не только область газообразного состояния вещества, но также области двухфазного и жидкого состояний. Жидкому состоянию соответствуют круто уходящие вверх левые участки изотерм. Однако в этой области имеется лишь качественное согласие с результатами экспериментов.
Сравнение изотерм Ван-дер - Ваальса ( рис. 12.7) с экспериментальными изотермами реальных веществ ( например, с рис. 12.5) показывает, что изотермы Ван-дер - Ваальса охватывают не только область газообразного состояния вещества, но также области двухфазного и жидкого состояний. Жидкому состоянию соответствуют круто уходящие вверх левые участки изотерм. Однако в этой области имеется лишь качественное согласие с результатами экспериментов.
КС
Критическое состояние
1) предельное состояние равновесия двухфазных систем, в котором обе сосуществующие фазы (См. Фаза) становятся тождественными по своим свойствам;
2) состояние вещества в точках фазовых переходов (См. Фазовый переход) II рода. К. с., являющееся предельным случаем равновесия двухфазных систем, наблюдается в чистых веществах при равновесии жидкость — газ, а в растворах — при фазовых равновесиях (См. Фазовое равновесие) газ — газ, жидкость — жидкость, жидкость — газ, твёрдое тело — твёрдое тело. На диаграммах состояния (См. Диаграмма состояния) К. с. соответствуют предельные точки на кривых равновесия фаз (рис. 1, а и б) — т. н. критические точки (См. Критическая точка). Согласно фаз правилу (См. Фаз правило) критическая точка изолирована в случае двухфазного равновесия чистого вещества, а, например, в случае бинарных (двойных) растворов (См. Растворы) критические точки образуют критическую кривую в пространстве термодинамических переменных (параметров состояния). Значения параметров состояния, соответствующие К. с., называются критическими — критическое давление рк, критическая температура Тк, критический объём Vк, критический состав хк и т. д.
С приближением к К. с. различия в плотности, составе и др. свойствах сосуществующих фаз, а также теплота фазового перехода и межфазное поверхностное натяжение уменьшаются и в критической точке равны нулю.
В том случае, когда кривая сосуществования фаз заканчивается критической точкой, оказывается принципиально возможным перевести вещество из одной фазы в другую, минуя область расслоения на две фазы (например, газ превратить в жидкость, изменяя его состояние по линии AB на рис. 1, а, т. е. минуя область, где одновременно существуют газ и жидкость). Сжижение (конденсацию) газов возможно осуществить лишь после их охлаждения до температур, меньших Тк.
В двухкомпонентных системах характерные для К. с. явления наблюдаются не только в критической точке равновесия жидкость — газ, но и в так называемых критических точках растворимости, где взаимная растворимость компонентов становится неограниченной. Существуют двойные жидкие системы как с одной, так и с двумя критическими точками растворимости — верхней и нижней (рис. 2, а и б). Эти точки являются температурными границами области расслаивания жидких смесей на фазы различного состава. Аналогичной способностью к расслаиванию при определённой критической температуре обладают некоторые растворы газов и Твёрдые растворы.
Переход системы из однофазного состояния в двухфазное вне критической точки и изменение состояния в самой критической точке происходят существенно различным образом. В первом случае при расслоении на две фазы переход начинается с появления (или исчезновения) бесконечно малого количества второй фазы с конечным отличием её свойств от свойств первой фазы, что сопровождается выделением или поглощением теплоты фазового перехода. Поскольку возникновение такой новой фазы приводит к появлению поверхности раздела и поверхностной энергии, для её рождения требуются достаточно большие зародыши. Это означает, что при таком фазовом переходе (фазовом переходе 1 рода) возможны переохлаждение или перегрев первой фазы, обусловленные отсутствием жизнеспособных зародышей новой фазы.
Фазовые переходы в критических точках, являющихся предельными на кривых равновесия фаз, представляют собой частные случаи фазовых переходов II рода. В критической точке фазовый переход происходит в масштабах всей системы. Флуктуационно возникающая новая фаза по своим свойствам бесконечно мало отличается от свойств исходной фазы. Поэтому возникновение новой фазы не связано с поверхностной энергией, т. е. исключается перегрев или переохлаждение, и фазовый переход не сопровождается выделением или поглощением теплоты и скачком удельного объёма (фазовый переход II рода).
При приближении к К. с. физические свойства вещества резко изменяются: теоретически неограниченно возрастает теплоёмкость и восприимчивость системы к внешним воздействиям (например, изотермическая сжимаемость в случае чистых жидкостей, магнитная восприимчивость у ферромагнетиков и т. д.); наблюдается целый ряд др. особенностей в поведении вещества (см. Критические явления). Эти особенности, характерные для К. с. объектов самой различной природы, объясняются тем, что свойства вещества в К. с. определяются не столько конкретными законами взаимодействия его частиц, сколько резким возрастанием в веществе флуктуаций и радиуса их корреляции. Знание особых свойств веществ в К. с. необходимо во многих областях науки и техники: при создании энергетических установок на сверхкритических параметрах, сверхпроводящих систем, установок для сжижения газов, разделения смесей и т. д.
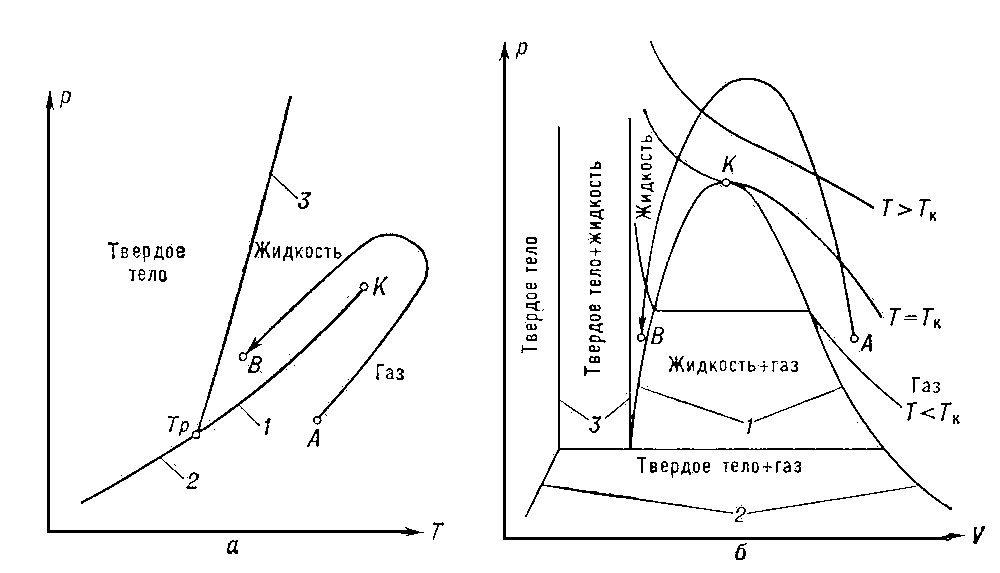
Рис. 1. а — диаграмма состояния чистого вещества в координатах р, Т. Кривые сосуществования обозначены цифрами: 1 — равновесие жидкость — газ, 2 — твёрдое тело; 3 — твёрдое тело — жидкость; К — критическая точка, Т = Тк — критическая изотерма; б — диаграмма в координатах р, V. Цифрами обозначены области сосуществования двух фаз: 1 — жидкость — газ; 2 — твёрдое тело — газ; 3 — твёрдое тело — жидкость.
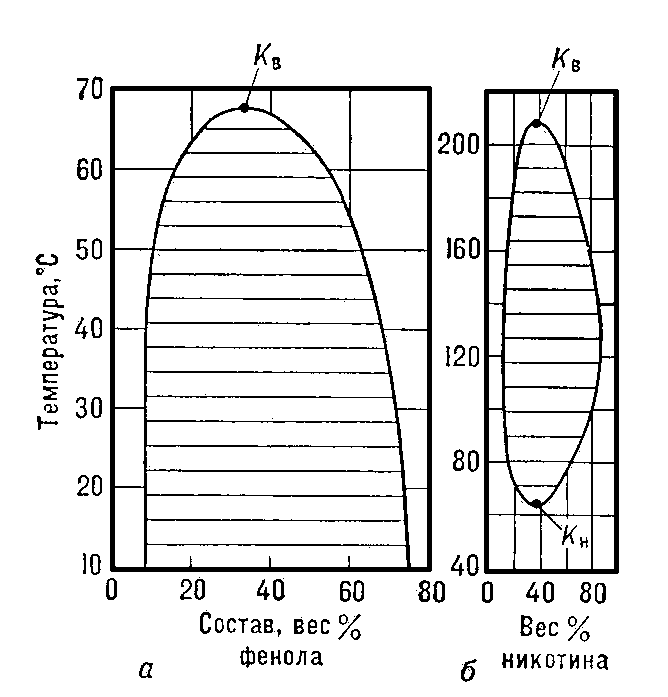
Рис. 2. а — верхняя критическая точка (Кв) жидкой смеси фенол — вода (Tк ≈ 66°С). Заштрихована область, где смесь состоит из двух фаз, имеющих различную концентрацию компонентов; б — двухкомпонентная жидкая система никотин — вода, имеющая как верхнюю критическую точку растворения (Кв с Tк = 208°С), так и нижнюю критическую точку (Кн с Тк ≈ 61° С).