

- •Preface
- •Acknowledgments
- •Contents
- •Contributors
- •1. Introduction
- •2. Evaluation of the Craniomaxillofacial Deformity Patient
- •3. Craniofacial Deformities: Review of Etiologies, Distribution, and Their Classification
- •4. Etiology of Skeletal Malocclusion
- •5. Etiology, Distribution, and Classification of Craniomaxillofacial Deformities: Traumatic Defects
- •6. Etiology, Distribution, and Classification of Craniomaxillofacial Deformities: Review of Nasal Deformities
- •7. Review of Benign Tumors of the Maxillofacial Region and Considerations for Bone Invasion
- •8. Oral Malignancies: Etiology, Distribution, and Basic Treatment Considerations
- •9. Craniomaxillofacial Bone Infections: Etiologies, Distributions, and Associated Defects
- •11. Craniomaxillofacial Bone Healing, Biomechanics, and Rigid Internal Fixation
- •12. Metal for Craniomaxillofacial Internal Fixation Implants and Its Physiological Implications
- •13. Bioresorbable Materials for Bone Fixation: Review of Biological Concepts and Mechanical Aspects
- •14. Advanced Bone Healing Concepts in Craniomaxillofacial Reconstructive and Corrective Bone Surgery
- •15. The ITI Dental Implant System
- •16. Localized Ridge Augmentation Using Guided Bone Regeneration in Deficient Implant Sites
- •17. The ITI Dental Implant System in Maxillofacial Applications
- •18. Maxillary Sinus Grafting and Osseointegration Surgery
- •19. Computerized Tomography and Its Use for Craniomaxillofacial Dental Implantology
- •20B. Atlas of Cases
- •21A. Prosthodontic Considerations in Dental Implant Restoration
- •21B. Overdenture Case Reports
- •22. AO/ASIF Mandibular Hardware
- •23. Aesthetic Considerations in Reconstructive and Corrective Craniomaxillofacial Bone Surgery
- •24. Considerations for Reconstruction of the Head and Neck Oncologic Patient
- •25. Autogenous Bone Grafts in Maxillofacial Reconstruction
- •26. Current Practice and Future Trends in Craniomaxillofacial Reconstructive and Corrective Microvascular Bone Surgery
- •27. Considerations in the Fixation of Bone Grafts for the Reconstruction of Mandibular Continuity Defects
- •28. Indications and Technical Considerations of Different Fibula Grafts
- •29. Soft Tissue Flaps for Coverage of Craniomaxillofacial Osseous Continuity Defects with or Without Bone Graft and Rigid Fixation
- •30. Mandibular Condyle Reconstruction with Free Costochondral Grafting
- •31. Microsurgical Reconstruction of Large Defects of the Maxilla, Midface, and Cranial Base
- •32. Condylar Prosthesis for the Replacement of the Mandibular Condyle
- •33. Problems Related to Mandibular Condylar Prosthesis
- •34. Reconstruction of Defects of the Mandibular Angle
- •35. Mandibular Body Reconstruction
- •36. Marginal Mandibulectomy
- •37. Reconstruction of Extensive Anterior Defects of the Mandible
- •38. Radiation Therapy and Considerations for Internal Fixation Devices
- •39. Management of Posttraumatic Osteomyelitis of the Mandible
- •40. Bilateral Maxillary Defects: THORP Plate Reconstruction with Removable Prosthesis
- •41. AO/ASIF Craniofacial Fixation System Hardware
- •43. Orbital Reconstruction
- •44. Nasal Reconstruction Using Bone Grafts and Rigid Internal Fixation
- •46. Orthognathic Examination
- •47. Considerations in Planning for Bimaxillary Surgery and the Implications of Rigid Internal Fixation
- •48. Reconstruction of Cleft Lip and Palate Osseous Defects and Deformities
- •49. Maxillary Osteotomies and Considerations for Rigid Internal Fixation
- •50. Mandibular Osteotomies and Considerations for Rigid Internal Fixation
- •51. Genioplasty Techniques and Considerations for Rigid Internal Fixation
- •52. Long-Term Stability of Maxillary and Mandibular Osteotomies with Rigid Internal Fixation
- •53. Le Fort II and Le Fort III Osteotomies for Midface Reconstruction and Considerations for Internal Fixation
- •54. Craniofacial Deformities: Introduction and Principles of Management
- •55. The Effects of Plate and Screw Fixation on the Growing Craniofacial Skeleton
- •56. Calvarial Bone Graft Harvesting Techniques: Considerations for Their Use with Rigid Fixation Techniques in the Craniomaxillofacial Region
- •57. Crouzon Syndrome: Basic Dysmorphology and Staging of Reconstruction
- •58. Hemifacial Microsomia
- •59. Orbital Hypertelorism: Surgical Management
- •60. Surgical Correction of the Apert Craniofacial Deformities
- •Index
58
Hemifacial Microsomia
John H. Phillips, Kevin Bush, and R. Bruce Ross
There is no other syndrome in the head and neck region that presents the craniofacial surgeon with such diverse choices for management as hemifacial microsomia. All patients require a multidisciplinary approach to management involving both conservative (nonsurgical) and surgical techniques. The variability of the condition generally called hemifacial microsomia has made it difficult to devise an accurate label for the condition. As Gorlin et al.1 state, “While there are no agreed upon minimal diagnostic criteria, the typical phenotype is characteristic when enough manifestations are present.”
Nomenclature
One of the areas concerning hemifacial microsomia is what the syndrome should be called. The most commonly used labels are oculo-auriculo-vertebral spectrum,2 or Goldenhar syndrome,3 a variation or subgroup of hemifacial microsomia.4,5 Other terms are commonly seen describing a spectrum of deformities that encompass auricular anomalies in combination with mandibular deformities and macrostomia are otomandibular dysostosis,6 first arch syndrome,7 first and second branchial arch syndrome,8 lateral facial dysplasia,4 and facio-auriculo-vertebral (FAV) malformation complex.5 Converse et al.9 coined the term craniofacial microsomia in recognition of the fact that there is approximately a 20% incidence of bilaterality in this syndrome.5
For the purpose of this review, we have chosen the term hemifacial microsomia (HFM) to represent a syndrome consisting of a constellation of deformities revolving around auricular deformities, craniofacial skeletal deformities (most notably the mandible and temporomandibular joint complex), and soft tissue deficiencies.
The most commonly affected structures are the external and middle ear, condyle and ramus of the mandible, muscles of mastication, parotid gland, zygomatic bone and arch, temporal bone, maxilla, and orbit. The abnormalities of the temporomandibular joint (TMJ) range from complete agenesis to subtle differences in form or size with few deformities. Invariably, however, the dysplasia is both a deficiency and a
malformation. Associated anomalies occur less frequently in the eye, vertebral column, and other parts of the body.
Epidemiology
Hemifacial microsomia is the second most common congenital craniofacial defect (after cleft lip and palate). Grabb estimated the frequency of HFM as 1 in 5,600 births.8 Other estimates of frequency range from 1 in 3,50010 to 1 in 26,550.11 Gorlin et al. believe the frequency is closer to Grabb’s estimate.1 There is a male to female ratio of 3:2 and also a 3:2 predilection for right-sided ear involvement.12,13 A significant feature is that in 70% of cases the condition appears to be unilateral. In bilateral cases, asymmetry is the rule, and rarely are both sides severely affected.
Differential Diagnosis
There are several syndromes and conditions with features that make confusion with hemifacial microsomia a possibility. The syndromes to consider in the differential diagnosis of HFM are Townes–Brocks syndrome, brachio-oto-rental (BOR) syndrome, mandibulofacial dysostosis (Treacher Collins), maxillofacial dysostosis, Rombergs, TMJ ankylosis (pathology or trauma), Nager acrofacial dysostosis, and acrofacial dysostosis.1
Etiology and Pathogenesis
Hemifacial microsomia is considered to be sporadic. Autosomal dominance transmission within a family has been documented.14 The syndrome is variable in its expression within those few families where genetic transmission is noted. HFM has a low recurrence risk (2%–3%),8,14 while frequently there is discordance in monozygotic twins. The mild forms of the condition are difficult to ascertain, so that an accurate familial history is virtually impossible to obtain. Genetic hetero-
727
728
geneity has been proposed to explain the variability in genetic transmission.
The defects in these cases appear to occur without relation to embryonic differentiation. One sees adjacent structures with the same origin but only one affected, and adjacent structures from completely different origins where both are affected. Pure unilateral dysplasias occur frequently. All these findings lead to the conclusion that in many cases the insult to the embryo occurs at a time when tissue differentiation is well advanced, possibly even completed on occasion, and that the injury is very localized.4,10,15,16 The mechanism may be a local injury that would cause cell destruction, interference with cell movement and differentiation, or displacement of areas of cells.
Two theories regarding the pathogenesis of HFM are currently discussed in the literature. Stark and Saunders suggested that Hoffstetter and Veau’s theory of mesodermal deficiency could be applied to hemifacial microsomia.7 Poswillo developed an animal model in which the induction of early vascular disruption and the subsequent expanding hematoma by in utero administration of triazene produced a phenotype that was very similar to hemifacial microsomia in the mouse.10 This and subsequent hematoma formation caused local destruction of tissue and delayed differentiation and induced abnormal development in adjacent structures. The hypothesis is attractive because of the high variability and asymmetry of HFM malformations. His findings provide an explanation for the great variability in the expression of these anomalies, because hemorrhages may vary in number, size, and location, and may occur simultaneously in other parts of the body. The specimens in Poswillo’s study, however, showed numerous abnormalities (e.g., of the brain) that are not typical of HFM, and there were severe abnormalities already present at the time the hemorrhage occurred (e.g., micrognathic). Newman and Hendricks17 repeated the Poswillo study and concluded that the resulting malformations were much more similar to Treacher Collins syndrome than hemifacial microsomia.
External and middle ear malformations commonly seen in the retinoic acid syndrome (RAS)18 are at least superficially similar to those of HFM. As these features of the RAS appear to be related to interference with neural crest development,19,20 such interference may be responsible for at least some HFM variants. Also, the cardiovascular outflow tract malformations sometimes noted in HFM cases are characteristic in RAS. Vertebral defects in Goldenhar syndrome are similar to those produced in mice by retinoic acid.21 The administration of thalidomide to monkeys has also produced an HFM phenotype by inducing hemorrhage at an early fetal stage.22 Kleinsasser and Schlothan have noted a significant number of newborns with first and second branchial arch abnormalities after administration of thalidomide during pregnancy.23 This has been considered presumptive evidence for the vascular hematoma theory because thalidomide is known to cause bleeding.
J.H. Phillips, K. Bush, and R.B. Ross
Clinical Manifestations
Hemifacial microsomia manifests itself in a diverse manner. Evidence of HFM can be seen not only throughout the affected facial skeleton but in other systems as well. Goldenhar syndrome makes up about 10% of all cases and is distinguished from hemifacial microsomia by the presence of epibulbar dermoids and vertebral anomalies (notably hemivertebrae).2 Only cursory mention is given here to anomalies outside the craniofacial region or those that do not directly impact upon the treatment regime. A detailed summary of anomalies that can be present in hemifacial microsomia is found in Gorlin et al.1
Neurologic
Several associated deformities can have direct impact on treatment regimes. Complete or partial paralysis of the facial nerve on the affected side is present in 10% to 20% of cases.8,24 The marginal mandibular branch is most often affected. The course of the facial nerve may be abnormal, and this should be kept in mind during temporomandibular reconstruction.25,26 Almost any cranial nerve can be affected, including the trigeminal.27 There is commonly a hypoplasia or paralysis of the ipsilateral tensor veli palatini8,25; therefore, on intraoral examination the soft palate deviates to the opposite side. Luce et al. found an incidence of one-third of patients presenting with moderate to severe hypernasality.28 Sprinzten et al.29 found 55% of HFM patients presenting with velopharyngeal insufficiency, and cleft lip or palate is present in 7% to 15% of cases.14,30
It is useful when assessing the patient with hemifacial microsomia to keep the following six areas in mind: cranial, orbital, midface, mandibular-temporomandibular joint complex, auricular, and soft tissue. Deformities are discussed here in this manner as it aids in clearly delineating the deformities and determining a coherent, chronologically organized treatment plan.
Cranial
Deformities in the cranial region tend to occur with more severe forms of HFM. A number of skull defects ranging from cranium bifidum to microcephaly and plagiocephaly have been described with hemifacial microsomia.1 The squamous temporal bone may be flattened. The mastoid air cells may exhibit decreased pneumatization as well as flattening of the mastoid process. The petrous portion of the temporal bone is usually spared. The frontal bone may be flattened, mimicking plagiocephaly.
Orbital
Ophthalmologic anomalies besides deformities of the bony orbital cavity exist in hemifacial microsomia. Commonly
58. Hemifacial Microsomia
noted ophthalmologic deformities include the presence of epibulbar dermoids, microphthalmos, colobomas, and, in 22% to 25%, ocular motility disorders.1,31 The bony orbital cavity may be small. The lateral orbital rim and inferior orbital rim on the affected side may be retruded. Vertical dystopias can also occur.
Midface
The zygoma may be hypoplastic or even absent in severe cases. As a result of zygomatic deformity, cheek prominence is decreased and often benefits from augmentation of some kind. The canthal-tragus line may be shortened.
The maxilla is also frequently affected in all three dimensions. Thus, the maxillary sinus and nasal cavity may be smaller on the affected side, the maxilla is frequently deficient in vertical height, retruded (more posterior), and narrower in width as well. These abnormalities are most likely a primary deficit, rather than the result of inhibition from the impinging mandible as is frequently hypothesized in the literature.32,33 While normal tooth eruption in both maxillary and mandibular arches can easily be inhibited by the restriction of a deficient mandibular ramus, there is little reason to believe that the basal bone in either arch will be affected. The upward cant of the maxillary plane is probably an intrinsic maxillary deficiency, while the cant of the occlusal plane is invariably related to inhibited dental eruption plus the frequent deficiency of the maxilla itself. This is an important consideration in treatment because it is most unlikely that a deficient basal maxilla will respond to treatment that merely removes the mandibular interference, even though the teeth will quickly erupt and tend to level the occlusal plane.
729
restrictions, so that the affected side merely rotates while the “normal” side advances. (2) The vertical canting of the mandibular and occlusal planes result partly from a deficiency in vertical height of the ramus or condyle and partly from decreased bulk of the bony muscle attachments (masseter and medial pterygoid). Because the muscles are severely hypoplastic or absent in severe cases, this vertical asymmetry is invariably present. As mentioned earlier, the maxilla is also frequently affected in vertical height, exaggerating the asymmetry. Even if the maxilla itself is normal, the abnormal mandibular position will inhibit the eruption of the mandibular and maxillary teeth on the affected side, causing a tilting of the occlusal plane.3 There is a transverse asymmetry, which is partly the medial displacement of the mandibular ramus and condyle and partly soft tissue muscle hypoplasia, often including a maxillary deficiency as well.
Dental Compensations
Teeth erupt from the jaws and are guided toward the opposing teeth by the forces they encounter in the oral cavity. Generally, these are forces generated by the lip, tongue, and cheek musculature. When jaws are poorly related to each other, the teeth are guided to achieve the best occlusion. Teeth erupt until they encounter resistance, normally the teeth in the opposing arch, but the lip, tongue, or an external force (e.g., thumb) can halt tooth eruption. In hemifacial microsomia, even gross malrelation of the jaws will not result in gross malocclusion: the teeth will generally meet in an adequate relationship. If the mandible is deficient on one side and prevented from achieving a normal vertical relationship with the maxilla, eruption of the teeth on the affected side is inhibited in both jaws.
Mandibular-Temporomandibular Joint Complex
Mandibular deformities and ear deformities are the hallmark of hemifacial microsomia. Deformities can range from minimal shape deformities of the condyle to complete absence of the affected ramus and condyle and much of the body. The affected condyle is always abnormal, and this is probably the only constant feature of HFM. The mandibular-temporo- mandibular deformity varies from a minimal deformity of the complex to a complete absence. Frequently there is a bony deformity of the squamous temporal bone, and the posterior wall of the glenoid fossa may be abnormal or absent.
The facial asymmetry in HFM is three dimensional. With regard to the mandible, there is (1) an inadequate anteroposterior vector to condylar size, causing the deviation of the chin toward the affected side. The position of the chin is a function of condylar height and vertical ramus anteroposterior length. As well, the mandible deviates on opening toward the affected side. Opening is normally accompanied by advancement of the condyles, but in these individuals the muscles that accomplish this (the lateral pterygoid muscles) are absent or hypoplastic and the condyle may be inhibited by soft tissue
Facial Growth
The bony asymmetries appear to be stable during growth of the child in virtually all cases, showing no clinical or cephalometric signs of improving or worsening except in very rare cases of each. In the infant it may be very difficult to detect the degree of asymmetry because the fat pads in the cheeks and the roundness of the face obscure asymmetry. What sometimes appears to be a worsening of the asymmetry with growth may be an illusion caused by the greater increase in height and depth of the lower face than in width, making the existing asymmetry increasingly more obvious, and creating the impression that the face is becoming more asymmetric. However, there are many authoritative-sounding statements in the literature to the effect that these cases worsen with growth. There is absolutely no evidence for such statements: on the contrary, the various objective analyses we use indicate that growth of the dysplastic ramus and condyle is quite exuberant and continues at or near the growth of the contralateral “normal” side. Facial asymmetry, including the orbits and maxilla, does not perceptibly change in these cases, nor does
730
the tilt of the occlusal and mandibular planes alter with growth. Unfortunately, the “worsening” hypothesis has now been widely accepted as fact: this has greatly influenced treatment methods.
Auricular
Many people consider that the spectrum of auricular deformities extends from simple ear tags to the presence of only a vestigial remnant of the ear. Meurman has classified ear deformities into three grades.34 Grade 1 is a slightly malformed ear that is smaller than a normal ear, grade 2 is vertical cartilaginous remnant with complete atresia of the ear canal, and grade 3 is only a small remnant of the original ear. The affected ear is often inferiorly positioned relative to the normal ear. The severity of ear deformity parallels the mandibular deformity and is not directly parallel with hearing function.35 Hearing function can be significantly impaired, which is a particular problem in the bilaterally affected individual. Middle ear structures may be absent or rudimentary in severe cases.
Soft Tissue
Macrostomia is a frequent finding in hemifacial microsomia, especially in Goldenhar syndrome. A definite soft tissue deformity has been identified in hemifacial microsomia. The temporalis muscle and other muscles of mastication, as well as the parotid and subcutaneous tissue, may all be involved. The degree of deficiency varies with the severity of the deformity. The amount of soft tissue deficit is often less than initially assessed. Any attempts at soft tissue augmentation should be delayed until the majority of bone reconstruction is complete.
J.H. Phillips, K. Bush, and R.B. Ross
Classification
There is almost as much confusion about the classification of HFM as there is about the nomenclature of the associated constellation of abnormalities known as hemifacial microsomia. Converse et al.9 stated that “the deformity in hemifacial microsomia varies in extent and degree.” They considered that classification was difficult because of the heterogeneity of the syndrome. Meurman in 1957 provided us with an easily applicable classification system of microtia based upon the assessment of 74 patients,34 as previously described. Pruzansky modified Meurman’s classification to preauricular anomalies, that is, ear tags, and applied this modification to 90 cases of hemifacial microsomia.36
Longacre et al. developed a classification that involved dividing patients into groupings of unilateral and bilateral microtia.37 Subsequently, each group was then subdivided into levels of facial deformity. Converse et al. stated that because of the heterogeneity of the syndrome no accurate classification system was available and each case must be reviewed individually.38
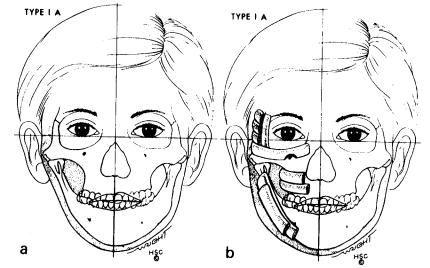
Most clinically successful classifications have revolved around the mandibular and temporomandibular skeletal deformity. Pruzansky described three grades of mandibular deformity.36 Each grade increases in severity until in grade 3 cases deformities may present with complete agenesis of the ramus. Swanson and Murray recognized the fact that the temporomandibular joint may be significantly deformed and acknowledged this in their classification of mandibular and temporomandibular deformities of HFM.39 In 1985, Lauritzen et al. classified mandibular-temporomandibular deformity into five grades of severity40 (Figures 58.1–58.6). The classification of Lauritzen et al. clearly focuses the craniofacial surgeon’s attention on the abnormalities of the craniofacial
FIGURE 58.1 (a) Hemifacial microsomia (HFM) type IA. Mandible is intact with horizontal occlusal plane. Contour augmentation only is needed. (b) Multiple onlay bone grafts of split rib have been added.

58. Hemifacial Microsomia |
731 |
FIGURE 58.2 (a) HFM type IB. Mandible is intact, but occlusal plane is tilted. Osteotomies are shown. (b) Postoperative skeletal alignment after a Le Fort I procedure, bilateral mandibular sagittal split, and a transposition genioplasty. A wedge of bone is grafted into the right maxilla.
FIGURE 58.3 (a) HFM type II. Mandible is incomplete with a deficient right ascending ramus. A sufficient glenoid fossa is present. (b) The ascending ramus of the mandible is constructed from a fullthickness costochondral graft.
FIGURE 58.4 (a) HFM type III. The right ascending ramus of the mandible is vestigial and the glenoid fossa is inadequate. (b) A transverse full-thickness rib graft is replacing the zygoma, and a TM joint is constructed.
732 |
J.H. Phillips, K. Bush, and R.B. Ross |
skeleton and hence an appropriate treatment plan for these anomalies.
In recognition of the fact that the abnormalities in hemifacial microsomia exist primarily in the facial skeleton, soft tissue, and auricle, several classification systems that identify abnormalities in all these areas have been developed. Two such classification systems are the OMENS41 and the SAT42 systems. SAT stands for skeletal, auricular, and soft tissue. The OMENS classification system described by Vento et al.41 is an acronym in which each letter stands for a major area of possible abnormality: O for orbital, M for mandibular, E for ear, N for nerve, and S for soft tissue. Each major area is further subdivided; that is, a modification of the Pruzansky classification is used in the M (mandibular) section. The SAT classification system is loosely based on the tumor node metastasis (TNM) tumor classification43 with subdivisions within each S, A, and T category. Both the OMENS and SAT
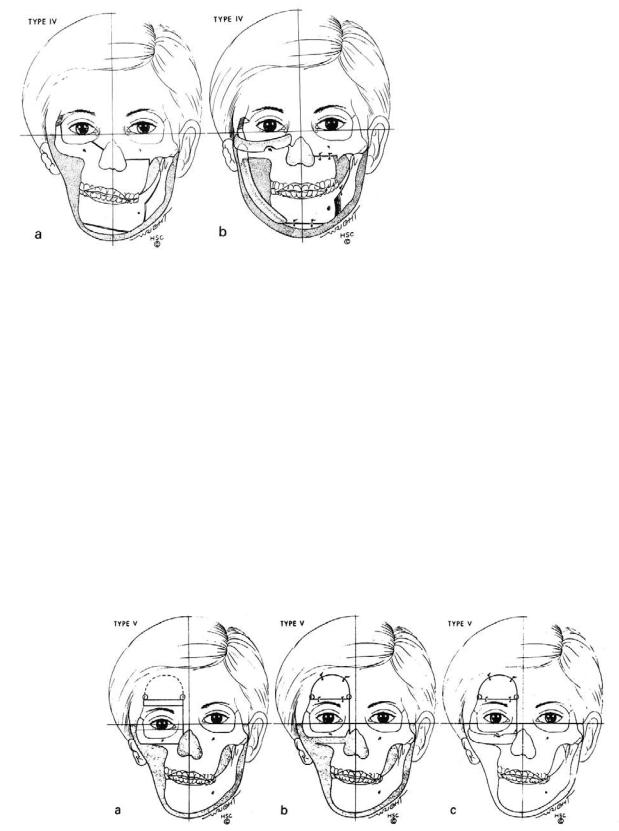
FIGURE 58.5 (a) HFM type IV. The right facial skeleton is retruded, and cuts for a right-sided Le Fort III and left-sided Le Fort I procedure are made. The right lateral orbital rim is cut obliquely so as to become self-retaining after transposition. (b) The facial skeleton is advanced, occlusal plane corrected, and mandible constructed.
classification systems are more complete than many other systems simply because of the one-dimensionality of many other systems. However, because of their complexity, both systems are somewhat unwieldy and we believe they fail to focus the attention of the treating physician on the relevant deformities requiring surgical intervention.
At the present time, our practice at The Hospital for Sick Children in Toronto is to classify hemifacial microsomia utilizing a combination of Meurman’s classification34 for congenital microtia and that of Lauritzen et al.40 for the skeletal deformities. Utilization of these two classification schemes focuses the surgeon’s attention on the anomalies requiring surgical intervention. The assessment of soft tissue deficiency should be postponed until after there has been skeletal reconstruction, as we believe it is difficult to estimate the soft tissue deficiency until the facial skeletal proportions are restored.
FIGURE 58.6 (a) HFM type V. Right orbit is dystopic. Cuts are planned including the craniotomy. (b) The right orbital box is moved upward and secured in place. The craniotomy is closed. (c) A zy-
gomatic arch is constructed from a full-thickness rib and the glenoid fossa is prepared. During the next stage operation, this patient will be treated as an HFM type II.
58. Hemifacial Microsomia
Treatment
The best approach to such a complex problem is a coordinated effort in which various specialists contribute their knowledge and skills to work together in planning and carrying out treatment customized to the specific needs of the patient. A craniofacial team might consist (in alphabetical order) of anaesthetist, audiologist, craniofacial surgeon, dentist, geneticist, ophthalmologist, orthodontist, otolaryngologist, pediatrician, plastic surgeon, psychiatrist, radiologist, social worker, and speech pathologist.
Indications for Treatment
Treatment would be easier if delayed until adolescence when facial growth has been essentially completed. The more stable structures permit quite precise surgical and orthodontic treatment planning, with the hope that a single surgery will be all that is necessary. Delaying surgery, however, subjects the child to living with a facial deformity through the most difficult years of social interaction and the development of self-esteem. Treatment of hemifacial microsomia can be divided into chronological time periods.
Age 0 to 5 Years
During this phase, complete assessment by the craniofacial team occurs. Problems such as feeding, speech, hearing, and genetic counseling are addressed. The only surgical interventions undertaken during this period are the correction of macrostomia, the removal of ear tags, correction of forehead deformities, and if the orbit is microphthalmic with no functional vision, the placement of an orbital expander.
“Plagiocephaly” or the retruded brow is addressed before 18 months of age using the surgical techniques developed for treating true plagiocephaly (unilateral coronal synostosis). Correction of the forehead deformity is undertaken before age 18 months to maximize bone formation after surgical intervention. It may be possible to correct some orbital dystopias at the same time as the forehead correction is done.
Orbital expansion is controversial. Our utilization of this technique is in individuals with no functional globe or vision in the affected eye. Placement of an expander in the affected globe before the age of 1 year is undertaken to try and simulate the normal growing globe, stimulate orbitozygomatic growth, and stretch the soft tissues around the eye.44,45 Although adequate growth may not occur with this technique in all individuals, those individuals who fail to grow can still be treated at a later date with orbital osteotomies or onlay bone grafting technique. The orbital expander is inflated with 0.5-ml increments weekly. Orbital volume changes are followed with intermittent computed tomography (CT) scans. Once appropriate volumes have been reached, the expander can be removed, and an orbital conformer manufactured to fit
733
the new orbital cavity is inserted at the same time as expander removal.
Age 5 to 8 Years
The main indication for early reconstructive surgery is to improve facial esthetics and provide the young child with a reasonably symmetric face through childhood, even though a second orthognathic surgery is frequently necessary at the conclusion of growth. Early treatment requires an evaluation of the positive or negative effects on the eventual result. With early mandibular surgery the teeth will adapt naturally to the new, more normal, relationship of the jaws. Dental compensations will be self-correcting, precluding the need for extensive reversal of long-established compensations, a difficult problem for the orthodontist if surgery is delayed until adolescence. A further advantage is that the soft tissues will grow and adapt to a more normal environment.
As a rule of thumb, then, in those mild cases in which the child and their family are not overly concerned with the facial esthetics, and there are no important functional indications, treatment should probably be delayed until adolescence. In such cases, soft tissue surgery or orthodontics alone may be all that is required to disguise the asymmetry. With the more severe deformities, extensive treatment in early childhood is usually indicated. Timing of treatment for moderate deformities demands good clinical judgment as well as evaluation of the child and the family.
During this phase of the child’s life, numerous surgical interventions may be planned such as auricular reconstruction, costochondral grafting, or temporomandibular joint reconstruction. Parents are advised of the options for ear reconstruction. Information on autogenous versus prosthetic ear reconstruction using implant technology is provided to the parents.46 If total ear reconstruction is warranted and an autogenous approach is adopted, the techniques for total ear reconstruction as popularized by Brent47 are utilized. It is our belief, if temporomandibular joint reconstruction, zygomatic reconstruction, or temporal bone augmentation are required, that total ear reconstruction should be performed after these surgical interventions so as to aid in correct positioning of the reconstructed ear. Our practice is not to reconstruct the middle ear on an individual with one normal ear and functional hearing. In the individual with bilateral involvement, ear reconstruction must be done in conjunction with middle ear reconstruction as dictated by the otolaryngologist member of the craniofacial team.
The surgical decision of no intervention, temporomandibular joint reconstruction, or costochondral grafting is based upon whether the individual has an adequate temporomandibular joint and an adequate ramus condyle complex. Utilizing the classification of Lauritzen et al., it is clear what skeletal surgical intervention is warranted during this period. Type IA has a level occlusal plane and requires only onlay bone grafting for cosmesis plus orthodontic intervention. The
734
temporomandibular joint and ramus are only slightly deformed. For types IB to V there is an occlusal tilt and malocclusion of a severity that warrants surgical intervention. The appropriate procedure is based upon the adequacy of the temporomandibular (TMJ)-mandibular complex. To try and maximize maxillary growth, and realizing that the risk of damage to permanent teeth is high if Le Fort I is used, the only surgical procedure performed at this age is costochondral grafting with TMJ reconstruction.
Type IB has a reasonable TMJ-mandibular complex and no surgical intervention is planned at this time. Bilateral sagittal split osteotomy, Le Fort I osteotomy, and genioplasty are planned for skeletal maturity.
Replacement of Defective TMJ
In the severe forms of hemifacial microsomia (types IIB and III), it is necessary to reconstruct the ramus and condyle and often create an articulating fossa as well. The major controversy in these cases is how to replace the missing tissues and the best time to begin surgical treatment. The method of choice is a bone graft to reconstruct the ramus, condyle, and temporomandibular joint. The most commonly used bone is a costochondral graft. The indications for costochondral grafting are the absence of a functional joint with consequent severe facial asymmetry.
Replacement of the defective TMJ for hemifacial microsomia is necessary to reconstruct the missing condyle and to create an articulating fossa as well. The method of choice is a bone graft to reconstruct the ramus, condyle, and temporomandibular joint. In looking at the need for a functional joint in the future with respect to the orthognathic surgery, the main requirement is buttressing of the most proximal portion of the ascending ramus or vestigial condyle with the base of the skull. This would prevent relapse of any sagittal split advancement and counterrotation, which is often required at skeletal maturity.
It is hard to determine the degree or actual interval absence of buttressing that is required to create the indication for costochondral grafting. In looking at a normal joint, it can be seen on the CT scan that there is often a 2- to 3-mm gap between the most proximal portion of the condyle in the cranial base. In this space, of course, there is the meniscus. At the present time, at our Center, if the gap between the most proximal portion of the mandible and cranial base is less than 10 mm and there is good mouth opening, then either nothing is done until orthognathic surgery at skeletal maturity or a sagittal split osteotomy may be done at 4 to 7 years of age to improve aesthetic appearance. It may be expected in some cases that a sagittal split advancement in the cases with less than 10 mm of bony gap may result in some relapse, necessitating a repeat sagittal split osteotomy. It is believed that the risk of redoing a sagittal split is less than the morbidity associated with a costochondral reconstruction.
In cases in which there is significant asymmetry and the
J.H. Phillips, K. Bush, and R.B. Ross
bony gap is greater than 10 mm, a costochondral graft may be indicated. If a joint is present, even with severe asymmetry, one would attempt to correct it by mandibular procedures such as a sagittal split or vertical ramus osteotomy, rather than the graft with its higher morbidity. In some cases, there is inadequate bone in the ramus for these procedures. Costochondral grafts are harvested with a periosteal sleeve from the contralateral side. The cartilage cap is shaped to form the condyle and then inserted via a combination of intraoral and preauricular incisions. We rigidly fix the costochondral graft to the vestigial ramus using lag screw techniques and a 1.5-mm plate as a washer. All patients are overcorrected with ipsilateral open bite (by as much as soft tissues will allow) at the time of surgery. Intermaxillary fixation is maintained for a period of 2 to 3 weeks to allow for comfort. Virtually all type II to V will benefit from orthognathic surgery at skeletal maturity to further level occlusion and aid in restoration of facial symmetry.
In types III to V, the zygomatic arch is absent or markedly hypoplastic and the glenoid fossa is nonexistent. It is these types of patients who should undergo glenoid reconstruction as outlined by Lauritzen et al. Our personal preference for glenoid fossa reconstruction and onlay bone grafting is to utilize split cranial bone grafts for reconstruction of the zygomatic arch and malar prominence. The condyle and ascending ramus is reconstructed with rib grafts. From age 5 years onward, the calvarium should be diploic and splittable, providing a good source of bone graft material. All bone grafts are fixed using currently available microand miniplate systems. The reconstructed glenoid fossa is lined with cartilage of the rib to increase the likelihood of a non-union.
Type V individuals pose special problems in that there is a significant orbital dystopia that will require osteotomies for correction. Our approach currently is to try and avoid type V patients by correcting some globe inadequacies by the use of an orbital expander. If osteotomy is required, we prefer to perform isolated zygomatic and orbital osteotomies and secondarily correct occlusion with a Le Fort I closer to skeletal maturity rather than utilize asymmetric Le Fort III and Le Fort I combinations during this time period of the child’s growth.
With the increasing use of bone-lengthening techniques, it will be interesting to note whether the proximal portion of the osteotomized mandible is driven toward the cranial base to create buttressing and, therefore, to obviate the need for costochondral graft reconstruction.
Costochondral Grafts
In recent studies at our Center,48 many factors were considered in estimating the success of a costochondral graft, including the etiology of the defect, surgical complexity of the procedure, age at surgery, previous surgery to the area, and the surgeon’s experience. Although there were minor differences, no factors except age at surgery were remarkable. The
58. Hemifacial Microsomia
early grafts were far more successful with gradually declining success until age 14. From age 3 to 9 years, the success rate of 19 grafts was 80%, while from 14 years onward the rate for 20 grafts fell to 50%. Although the difference was not quite statistically significant ( 2, p 0.06), it does seem that early placement of a costochondral graft is more successful.
One of the problems with early grafting is the different growth rates of the mandible and the graft.49,50 The otherwise successful graft may grow at a different rate than the contralateral natural condyle. The Ross 1996 study of the long-term followup of 13 grafts in growing children showed that subsequent growth of the graft was equal to that of the “normal” side in 6 cases, less in 2 cases, and greater in 5 cases.51
A possible explanation for this may lie with the size of the germinative zone of cartilage in the graft. The prechondrocytes in this zone supply cells for the proliferative zone, where interstitial growth is responsible for increased length of cartilage. Peltomaki and Ronning52 have shown that when costochondral grafts were transplanted to a nonfunctional area in rats, the growth in length of the graft varied with the thickness of this zone of cells. Removal or injury to these cells inhibited growth. Clinical control of the amount of graft growth may be possible if these findings could be adjusted to the need. They also showed53 that mature, nongrowing ribs transferred to a nonfunctional area in growing rats grew significantly. Their findings indicate a systemic, hormonal stimulation rather than a functional one.
Orthodontic Treatment
There are two conflicting theories with regard to the indications for and efficacy of orthodontic treatment in hemifacial microsomia. The first, held by most experienced clinicians, is that orthodontic treatment is not effective in producing meaningful change in the dysmorphic structures present in this condition. Rather, orthodontic mechanics and forces affect the teeth and associated alveolar process, but do not affect the underlying basal bone or condylar growth except in a limited, clinically insignificant degree if at all. Thus, orthodontic treatment in hemifacial microsomia is confined to alignment of the teeth in preparation for surgery and subsequent finishing procedures.
The second approach, pursued by Harvold and his followers,54 is that functional appliances will stimulate growth of the defective condyle to a meaningful degree. It is based on the mistaken belief that little growth occurs naturally and that the deficiency will worsen with growth. Their treatment consists of the wearing of functional appliances throughout childhood (providing further esthetic and psychic trauma) with surgery to the mandible in adolescence. For the Harvoldians, growth subsequent to appliance wear is proof of the efficacy of their treatment, when in fact growth would have occurred without their intervention (as explained earlier). These appli-
735
ances are unsuccessful in increasing mandibular sagittal growth. Evidence is very flimsy that these appliances ever work, let alone with consistency, in HFM. Harvold also claimed that a functional appliance must be worn before surgery to prepare the tissues and after surgery to maintain the graft. These procedures have been shown to the unnecessary in our clinic.
There is no question that the occlusal plane can be altered by changing the oral environment, either by directly lowering (distracting) the mandible by surgery, or by holding the mandible open with a unilateral bite pad that inhibits the teeth on the ‘normal’ side and allows the teeth on the affected side to erupt, thus leveling the occlusal plane. This does not, of course, affect mandibular symmetry in length or vertical height. In mild or even moderate cases (types I and IIA), the effect of the bite pad therapy is satisfactory and may avoid the need for maxillary surgery. In more severe cases in which the basal maxilla is asymmetric (as is the case in approximately one-third of individuals), however, the cant of the lips and the incisor teeth will not be perceptibly altered by this treatment, so facial esthetics are rarely improved.
Orthodontic treatment plays an important role in the preparation for surgery for the correction of facial asymmetry. Presurgically, dental arch alignment removes interferences that would prevent the mandible from being precisely positioned during surgery. After surgery an open bite usually appears on the affected side. Once the fixation wires are removed, the splint is often used to allow the extrusion or eruption of the maxillary and mandibular teeth. When the maxillary teeth are in contact with the mandibular teeth, the splint may be removed and braces applied to interdigitate the teeth.
Psychosocial
An important element of management of hemifacial microsomia is the monitoring of psychosocial adjustment. The major concern is the child’s future self-esteem and social competence. Parents are reassured that the severity of the craniofacial malformation is much less important than the strength of the family in determining how the child will ultimately adjust and succeed in life. The psychosocial team can monitor the patient’s self-esteem as treatment proceeds through childhood and adolescence and encourage the development of particular skills and talents.
Specific concerns include adjustment to body image and general self-concept as well as ability to relate to family, peers, and strangers. Motivation for and expectations of surgery should be carefully explored. Play therapy can be used to help the child integrate the experiences of hospitalization and surgery, and individual therapy is available for adolescents. For young children, “fitting in” becomes more important than pleasing parents. In the primary grades, children who are different from their peers often have some difficulty. They may be teased and called names. Some children learn to cope
736
by educating their classmates. Others rely on their personality strengths such as a quick wit or ability to achieve or by demonstrating specific talents. Self-esteem is determined primarily by the ability to feel genuinely positive about one’s self or some aspect of one’s life, whether it be academic success, sports, music, art, or a particular hobby.
During the early school years, many parents are particularly concerned that their child be educated in an environment without pity, overprotection, or underestimation of potential. Because teachers are largely unfamiliar with craniofacial problems, it is helpful if parents explain their child’s condition. There is a difficult balance to be found between protecting the child from the cruelty of teasing, stares, and questions and letting them cope with the world as it is and build ego strengths.
As the child reaches puberty, appealing to the opposite sex becomes important. Virtually all teenagers go through periods of doubt and insecurity. It is not surprising, therefore, that teenagers with a facial defect experience a sharp decline in self-confidence. They become more aware of the impact the facial problems may have on their lives, and also of the limitations of treatment. It is a very difficult period but most manage to cope effectively, especially if they have developed areas of competence unrelated to appearance. Some appear indifferent to the opposite sex and focus on academic or athletic achievements. Life usually becomes much easier when their peers mature and learn to see the person behind the facial appearance.
References
1.Gorlin RJ, Cohen MM Jr, Levin LS. Syndromes of the Head and Neck. New York: Oxford University Press; 1990.
2.Gorlin RJ, Jue KL, Jacobson NP, et al. Oculoauriculovertebral dysplasia. J Pediatr. 1963;63:991–999.
3.Goldenhar M. Associations malformatives de l'oeil et de l’oreille, en particulier le syndrome dermoide epibulbaire-appen- dices auriculaires-fistula auris congenita et ses relations avec la dysostose mandibulofaciale. J Genet Hum. 1952;1:243–282.
4.Ross RB. Lateral facial dysplasia (first and second branchial arch syndrome), hemifacial microsomia. Birth Defects Orig Artic Ser. 1975;11:51–59.
5.Smith DW. Facio-Auriculo-Vertebral Spectrum, in Recognizable Patterns of Human Malformations. 3rd ed. Philadelphia: WB Saunders; 1982:497–500.
6.Francois JJ, Haustrate L. Anomalies colomateuses du globe oculaire et syndrome du premier arc. Ann Ocul. 1954;187:340–368.
7.Stark RB, Saunders DE. The first branchial syndrome: the oral mandibular-auricular syndrome. Plast Reconstr Surg. 1962;29: 229–239.
8.Grabb WC. The first and second branchial arch syndrome. Plast Reconstr Surg. 1965;36:485–508.
9.Converse JM, Coccaro PJ, Becker H, Wood-Smith D. Clinical aspects of craniofacial microsomia. In: Converse JM, McCarthy JG, Wood-Smith D, eds. Symposium on Diagnosis and Treatment of Craniofacial Anomalies. St. Louis: CV Mosby; 1979: 461–475.
J.H. Phillips, K. Bush, and R.B. Ross
10.Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973;35:302–328.
11.Melnick M. The etiology of external ear malformations and its relation to abnormalities of the middle ear, inner ear, and other organ systems. Birth Defects Orig Artic Ser. 1980;16:303–331.
12.Coccaro PJ, Becker MH, Converse JM. Clinical and radiographic variations in hemifacial microsomia (review). Birth Defects Orig Artic Ser. 1975;11:314–324.
13.Rollnick BR, Kaye CI, Nagatoshi K, et al. Oculoauriculovertebral dysplasia and variants: phenotypic characteristics of 294 patients. Am J Med Genet. 1987;26:361–375.
14.Hermann J, Opitz JM. A dominantly inherited first arch syndrome. First conference on clinical delineation of birth defects. Part II. Malformation syndromes. In: Bergsma D, ed. Birth Defects, Original Article Series. Vol 2, No 2. New York: National Founda- tion–March of Dimes/Baltimore: Williams & Wilkins; 1969.
15.Ross RB, Johnston MC. Developmental anomalies and dysfunctions of the temporomandibular joint. In: Zarb GA, Carlson EE, eds. Temporomandibular Joint: Functions and Dysfunction.
Copenhagen: Munksgaard; 1994.
16.Johnston MC. The neural crest in abnormalities of the face and brain. Birth Defects. 1975;7:1–18.
17.Newman LM, Hendricks AG. Fetal ear malformations induced by maternal ingestion of thalidomide in the bonnet monkey (Macaca radiata). Teratology. 1981;23:351–364.
18.Lammer EJ, Chen DT, Hoar RM, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837–847.
19.Goulding EH, Pratt RM. Isoretinoin keratogenicity in mouse whole embryo culture. J Craniofacial Genet Dev Biol. 1986;6: 99–112.
20.Webster WS, Johnston MC, Lammer EJ, et al. Isoretinoin embryopathy and the cranial neural crest: an in vivo and in vitro study. J Craniofacial Genet Dev Biol. 1986;6:211–222.
21.Jarvis BL, Johnston MC, Sulik KK. Congenital malformations of the external, middle and inner ear produced by isoretinoin exposure in mouse embryos. Otolaryngol Head Neck Surg.
1990;102:391–401.
22.Poswillo D. Hemorrhage in the development of the face. Birth Defects. 1975;11(7):61–81.
23.Kleinsasser O, Schlothan R. Die Ohrmissbildungen im Rahmen der Thalidomid-Embryopathie. Z Laryngol Rhinol Otol. 1964; 43:344.
24.Basilla MK, Goldenberg R. The association of facial palsy and/or sensorineural hearing loss in patients with hemifacial microsomia. Am J Med Genet. 1989;26:287–291.
25.Converse JM, Coccaco PJ, Becker M, Wood-Smith D. On hemifacial microsomia. The first and second branchial arch syndrome. Plast Reconstr Surg. 1973;51:268–279.
26.Yovich J, Mulcahy M, Patson P. IVF and Goldenhar syndrome (letter). J Med Genet. 1987;24:644.
27.Aleksic S, Budzilovich G, Greco MA, et al. Intracranial lipomas, hydrocephalus and other CNS anomalies in oculoauriculo-ver- tebral dysplasia (Goldenhar-Gorlin syndrome). Child’s Brain. 1984;11:285–297.
28.Luce EA, McGibbon B, Hoopes JE. Velopharyngeal insufficiency in hemifacial microsomia. Plast Reconstr Surg. 1977;60: 602–606.
29.Sprintzen RJ, Croft CB, Berkman MD, Rakoff SJ. Velopharyngeal insufficiency in the facio-auriculo-vertebral malformation complex. Cleft Palate J. 1980;17:132–137.
58. Hemifacial Microsomia
30.Feingold M, Baum J. Goldenhar’s syndrome. Am J Dis Child. 1978;132:136–138.
31.Hertle RW, Quinn GE, Katowitz JA. Ocular and adnexal findings in patients with facial microsomias. Ophthalmology. 1992;99(1):114–119.
32.Kaban LB, Moses MH, Mulliken JB. Correction of hemifacial microsomia in the growing child: a follow-up study. Cleft Plate
J.1986;23(suppl 1):50–52.
33.Moses MH, Kaban LB, Mulliken JB, et al. Facial growth after early correction of hemifacial microsomia. Presented at the 6th Annual Meeting of the American Association of Plastic Surgeons. San Diego, CA, May 1, 1985.
34.Meurman Y. Cogenital microtia and meatal atresia. Arch Otolaryngol. 1957, 66:443–463.
35.Caldarelli DD, Hutchinson JG Jr, Pruzansky S, Valvassori GE. A comparison of microtia and temporal bone anomalies in hemifacial microsomia and mandibulofacial dysostosis. Cleft Palate
J.1980;17:103–110.
36.Pruzansky S. Not all dwarfed mandibles are alike. Birth Defects. 1969;5:120–129.
37.Longacre JJ, DeStefano GA, Holmstand KE. Surgical management of first and second branchial arch syndromes. Plast Reconstr Surg. 1963;31:507–520.
38.Converse JM, Wood-Smith D, McCarthy JG, et al. Bilateral facial microsomia. Diagnosis, classification, treatment. Plast Reconstr Surg. 1974;54:413–423.
39.Swanson LT, Murray JE. Asymmetries of the lower part of the face. In: Whitaker LA, Randall P, eds. Symposium on Reconstruction of Jaw Deformities. St. Louis: CV Mosby; 1978:7.
40.Lauritzen C, Munro IR, Ross RB. Classification and treatment of hemifacial microsomia. Scand J Plast Reconstr Surg.
1985;19:33–39.
41.Vento AR, LaBrie RA, Mulliken JB. The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate-Craniofacial J. 1991;28:68–76 (discussion 77).
42.David DJ, Mahatumarat C, Cooter RD. Hemifacial microsomia:
737
a multisystem classification. Plast Reconstr Surg. 1987;80: 525–535.
43.Copeland MM. American joint committee on cancer staging and end results reporting: objectives and progress. Cancer (Phila). 1965;18:1637–1640.
44.Cepela MA, Nunery WR, Martin RT. Stimulation of orbital growth by the use of expandable implants in the anophthalmic cat orbit. Opthalmic Plast Reconstr Surg. 1992;8:157–167.
45.Eppley BL, Holley S, Sadove AM. Experimental effects of intraorbital tissue expansion on orbitomaxillary growth in anophthalmos. Ann Plast Surg. 1993;31:19–26.
46.Tjellstrom A, Hakansson B. The bone-anchored hearing aid. Design principles, indications, and long-term clinical results. Otolaryngol Clin North Am. 1995;28:53–72.
47.Brent B. Auricular repair with autogenous rib cartilages: two decades of experience with 600 cases. Plast Reconstr Surg. 1992;90:355–374.
48.Munro IR, Phillips JH, Griffin G. Growth after construction of the temporomandibular joint in children with hemifacial microsomia. Cleft Palate J. 1987;26:303–311.
49.Ware WH. Growth centre transplantation in temporomandibular joint surgery. Trans Int Conf Oral. 1970;148–157.
50.Ware WH, Brown SL. Growth centre transplantation to replace mandibular condyles. J Maxillofac Surg. 1981;9:50–58.
51.Ross RB. Costochondral gafts replacing the mandibular condyle.
Cleft Palate Craniofac J. 1999;36:334–349.
52.Peltomaki T, Ronning O. Interrelationship between size and tissue separating potential of costochondral transplants. Eur J Orthod. 1991;13:459–465.
53.Peltomaki T, Ronning O. Growth of costochondral fragments transplanted from mature to young isogeneic rats. Cleft Palate Craniofacial J. 1993;30:159–163.
54.Harvold EP. The theoretical basis for the treatment of hemifacial microsomia. In: Harvold EP, Vargervik K, Chierici G, eds. Treatment of Hemifacial Microsomia. New York: AR Liss;1983.