

Колебательные спектры
Поглощение в области 102 – 103 см-1 (ИК – область) обусловлено обычно колебательными переходами при неизменном электронном состоянии молекулы; соответствующие спектры называют колебательными. Точнее их следовало бы назвать колебательно-вращательными, т. к. изменение колебательной энергии молекулы при поглощении в этой области сопровождается, как правило, изменением и вращательной энергии.
h = Е′ – Е″ = (Eвр′ + Екол′) – (Eвр″ + Екол″) . (2.104)
Колебательный спектр состоит из ряда довольно далеко отстоящих друг от друга полос, интенсивность которых с ростом волнового числа резко убывает (рис. 2.22). Первую, наиболее интенсивную полосу называют основной полосой, или основным тоном. Далее располагаются 1-й и 2-й обертоны. Интенсивность последующих полос убывает настолько резко, что уже 3-й и 4-й обертоны для большинства молекул наблюдать не удается.

Каждая полоса в спектре является сложной и при записи на приборе с большой разрешающей способностью прибора распадается на ряд отдельных линий. Появление такой тонкой структуры характерно для веществ в газообразном состоянии. Положение полос в спектре определяется колебательными переходами, а тонкая структура каждой полосы – вращательными переходами.
Для того чтобы понять происхождение такого спектра, рассмотрим вначале только колебательное движение и колебательные переходы, абстрагируясь от вращения молекул, т. е. примем
h = Екол′– Екол″ . (2.105)
Колебательное движение двухатомной молекулы с точки зрения классической механики можно представить как периодическое изменение расстояния между ядрами.

Согласно закону Гука, описывающему гармонические колебания, сила, возвращающая ядра в положение равновесия, пропорциональна смещению ядер из положения равновесия:
f = – kq , (2.106)
где k – силовая постоянная;
q – колебательная координата; q = ra + rb = r – re.
Уравнение Гука справедливо только для малых смещений ядер, т. е. когда q >> re, в пределе при q = 0.
Силовая постоянная двухатомной молекулы есть величина, характеризующая упругость связи и численно равная силе, формирующей (растягивающей или сжимающей) связь на единицу длины f = k при q = 1.
Элементарная работа упругой силы:
dA = – f dq . (2.107)
С учетом уравнения (2.106) получаем:
dA = – kq dq . (2.108)
После интегрирования в пределах
![]() (2.109)
(2.109)
для потенциальной энергии двухатомной молекулы получаем:
u = A = 1/2 kq2 . (2.110)
Из уравнения (2.110) следует, что
k = (d2u / dq2)q=0 . (2.111)
Таким образом, для малых смещений потенциальная энергия является квадратичной функцией от q = r – re. Кривая u–q или u–r – парабола, а силовая постоянная k характеризует кривизну параболы вблизи минимума.
При подстановке выражения (2.110) в уравнение Шредингера
2 кол + (8 2 / h2) (Екол – u) кол = 0 (2.112)
и решении этого уравнения получается следующее уравнение для собственных значений колебательной энергии двухатомной молекулы как гармонического осциллятора:
Екол = hо (v + 1/2) , (2.113)
где v – колебательное квантовое число, принимающее значения целых положительных чисел, начиная с нуля (v = 0, 1, 2, 3.......);
0 – собственная частота колебания вибратора.
Уравнение (2.113) можно представить в другом виде:
Екол = hce (v + 1/2) , (2.114)
где e – собственное волновое число (колебательная постоянная), характеризующее частоту колебаний, отнесенную к минимуму потенциальной кривой, т. е. ту частоту, которую согласно классической механике имела бы молекула для бесконечно малой амплитуды колебаний (q = 0, r = re). Величина e выражается в м-1 или см-1. Она является молекулярной постоянной. Любая двухатомная молекула характеризуется в каждом электронном состоянии некоторым постоянным значением e.
Уравнение (2.114) указывает на квантование колебательной энергии и на существование нулевой энергии осциллятора при v = 0:
Е0 кол = hce /2. (2.115)
Эта энергия не равна нулю. Энергия колебаний гармонического осциллятора возрастает прямо пропорционально квантовому числу v, что соответствует системе равноотстоящих квантовых уровней. Согласно квантово-механическим правилам отбора для гармонического осциллятора возможны переходы с v = 1. При поглощении света v изменяется на +1, увеличивается энергия и амплитуда колебаний.
Однако, модель гармонического осциллятора приводит к положениям, противоречащим экспериментальным данным:
1) Екол в рамках этой модели может быть сколь угодно большой. В этом случае химическая связь в молекуле была бы бесконечно упругой и ее разрыв был бы невозможен. Мы знаем, что это не так;
2) для гармонического осциллятора в спектре поглощения должна наблюдаться только одна полоса, что вытекает из правил отбора и эквивалентности колебательных уровней (рис. 2.23 а). Однако в спектре реальной двухатомной молекулы наблюдается несколько полос.
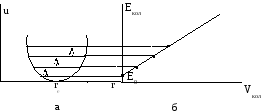
Рис. 2.23 Кривые потенциальной энергии (a) и зависимость колебательной энергии Екол от Vкол (б) для гармонического осциллятора
Все это означает, что реальные молекулы не являются гармоническими осцилляторами. Гармоническое приближение для них можно использовать только при малых смещениях ядер от положения равновесия, т.е. при малых значениях колебательного квантового числа (v = 0; 1).
Для реальных двухатомных молекул функция U(r) не является параболой, а возвращающая сила не строго пропорциональна величине смещения ядер. Это приводит к модели ангармонического осциллятора, для которого кривая потенциальной энергии изображается, как показано на рис. 2.24.
Для приближенного описания кривой потенциальной энергии используют функцию Морзе:
u = De [1 – e– (r – re)]2, (2.116)
где De – энергия диссоциации;
– постоянная для данной молекулы.

Рис. 2.24 Кривые потенциальной энергии (а) и зависимость колебательной энергии Екол от Vкол (б) для ангармонического осциллятора
При решении уравнения Шредингера для двухатомной молекулы, когда u(r) выражается функцией Морзе, собственные значения колебательной энергии Екол описываются двучленом:
Екол = hce (v +1/2) – hcexe (v + 1/2)2 , (2.117)
где хe – коэффициент ангармоничности, характеризующий отклонение от гармоничности, эта величина безразмерная, причем
e >> exe > 0. (2.118)
Из уравнения (2.117) можно получить выражение для нулевой энергии ангармонического осциллятора (где v = 0):
Е0 = 1/2 hce – 1/4 hcexe . (2.119)
Из уравнения (2.117) следуют выводы:
зависимость Екол от v не является линейной;
колебательные квантовые уровни сходятся при увеличении v.
Действительно, разность энергии колебаний при возрастании квантового числа на единицу уменьшается с ростом V:
Еv+1 v = E(v + 1) – E(v) = hc [e – 2exe (v+1)] . (2.120)
Найдем первую и вторую производные от функции (2.117):
Ev = hce (V + 1/2) – hcexe (V + 1/2)2 , (2.121)
dEV/dV = hc [e – 2exe (V + 1/2)] , (2.122)
d2EV/dV = –2hcexe < 0 . (2.123)
Выражение свидетельствует о том, что кривая Еv–V имеет максимум (рис 2.16,б) и колебательные уровни сходятся к некоторому значению Vмакс., которое можно найти из условия максимума:
dEV/dV = 0 , (2.124)
dEV/dV = hc[e – 2exe (Vмакс + 1/2)] = 0 , (2.125)
откуда
Vмакс = (e/2exe) – 1/2 , (2.126)
Vмакс
= 1/2xe
– 1/2
Таким образом, существует конечное число дискретных колебательных уровней и максимальная энергия ангармонического осциллятора ЕV,макс. Если молекуле сообщить колебательную энергию ЕV > EV,макс, произойдет диссоциация, как это видно из кривой потенциальной энергии (рис. 2.16,а).
Рассчитанные по формуле (2.127) значения Vмакс для большинства молекул составляет несколько десятков, для некоторых – до полутора сотен.
Правила отбора:
если для гармонического колебания осциллятора V = 1, то для ангармонического осциллятора квантово-механические правила отбора разрешает любые переходы: V = 1, 2, 3 и т. д.;
могут быть описаны любые вещества (полярные и неполярные).
Подставляя значения V, e, xe в уравнение (2.117) можно составить схему дозволенных уровней энергии колебания.
0
V=1
V=2 V=3
Е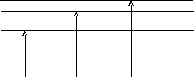

V=0
V=1
V=2
V=3
Рис. 2.25 Схема дозволенных уровней энергий колебаний.
Для большинства двухатомных молекул колебательный переход 01 требует 10 – 100 кДж/моль. Это значительно больше средней энергии теплового движения молекул газа при температуре 18 – 25оС (RT = 2,5 кДж/моль при 298оК). Поэтому можно считать, что при температуре опыта подавляющее большинство молекул находится на нижнем энергетическом уровне, т. е. V″=0.
Правило отбора позволяет вывести уравнение для всех частот, наблюдаемых в спектре и вывести колебательный спектр:
= EV/hc = e (V + 1/2) – exe (V + 1/2)2 . (2.128)
Подставляя величины V′ и V″ в уравнение (2.128) и беря разность волновых чисел получим:
V″0 = [e (V′ + 1/2) – exe (V' + 1/2)2] – [e (V″ + 1/2) – exe(V″ + 1/2)2]. (2.129)
После преобразования:
= (V' – V″) [e – exe (1 + V' + V″)] . (2.130)
Учитывая, что V’’=0, получим выражение для волновых чисел единственно экспериментально наблюдаемой серии переходов, показанных на рисунке, а именно переходов V″ (0)V':
= V' [e – exe (1+V')] , (2.131)
где V' = 1, 2, 3,..... Vмакс.
Наименьшая энергия требуется для перехода 01. Это соответствует появлению в спектре поглощения первой (низкочастотной) полосы – основной полосы, или основного типа. Переходы 02; 03 и т. д. дают последующие полосы – обертоны.
Волновые числа основной полосы и обертонов определяется в соответствии с (2.131) следующим образом:
01 основная полоса или обертон,
01 = e – 2exe = e (1 – 2xe), (2.132)
02 1-й обертон,
02 = 2e – 6exe = 2e (1 – 3xe), (2.133)
03 2-й обертон,
03 = 3e – 12exe = 3e (1 – 4xe), (2.134)
и т. д.
В общем случае для перехода 0V':
0V’ = V'e – V’(V’+1)exe . (2.135)
Из полученных выражений следует, что полосы поглощения в колебательном спектре сходятся, хотя, из-за того, что exe << e, эта сходимость для первых двух-трех полос выражена слабо. Величина exe составляет обычно несколько см-1, реже – десятки см-1, в то время как e = 102 – 103 см-1.
Вероятность перехода 01 наибольшая, чем и объясняется интенсивность основной полосы поглощения. Вероятность переходов 02; 03 и т. д. резко убывает с ростом V', что и отражается в спектре поглощения.
Определение колебательной постоянной e и коэффициента ангармоничности xe.
Важнейший результат экспериментального изучения ИК спектров поглощения состоит в определении молекулярных постоянных – колебательной постоянной e и коэффициента ангармоничности xe.
относят полосы поглощения к определенным колебательным переходам.
определяют частоту колебаний каждого перехода: 1, 2, 3.
сопоставляют каждой из частот уравнения типа (2.132) – (2.135) и, решив их совместно, определяют e и xe. Например:
 01
= e
(1–2xe)
01
= e
(1–2xe)
02 = 2e (1–3xe).
Определение энергии диссоциации (химической связи). Энергия химической связи есть та энергия, которую необходимо затратить, чтобы перевести молекулу с нулевого на максимальный колебательный квантовый уровень:
Напомним уравнение (2.127):
Vмакс = 1/2xe – 1/2 .
Подставив это уравнение в (2.127), получим:
E0Vmax = hce (1/2xe – 1/2 + 1/2) – hcexe (1/2x – 1/2 + 1/2)2 , (2.136)
E0Vmax = hce/2xe – hcexe/4xe = hcexe/4x, (2.137)
Emax = hce/4xe . (2.138)
Перейдем к мольным величинам энергии в Дж/моль:
Emax (моль) = Emax NА , (2.139)
Emax (моль) = hce NА /4xe . (2.140)
Энергию диссоциации, отсчитываемую от нулевого уровня и отнесенную к 1молю называют истинной энергией диссоциации и обозначают Dо:
Eх.с. = Do = Emax – E0 . (2.141)
Если энергию диссоциации отсчитывать от минимума потенциальной кривой, то она превышает D0 на величину нулевой энергии (рис. 2.18):
De = D0 + E0 . (2.142)
hceNА
4xe
hce
4xe
Напомним, что
Е0 = 1/2 hce – 1/4 hcexe ,
Тогда
D0 = hce/4xe – (hce/2 – hcexe/4) , (2.143)
D0 = (1–xe)2 . (2.144)
Переходя к мольным величинам, находим значение D0 в Дж/моль:
D0 = (1–xe)2. (2.145)
Таким образом: из колебательного спектра можно получить следующие молекулярные константы:
- собственную частоту колебаний е;
- коэффициент ангармоничности хе;
- энергию колебательного движения молекул;
- энергию химической связи.
Электронные спектры (основные понятия). При возбуждении электронов в молекулах наблюдается излучение в ультрафиолетовой и видимой областях спектра.
h = E'' – E' = (E''вр – E'вр) + (E''кол – E'кол) + (E''эл – E'эл).
П ри
этом имеет место совокупность всех
видов энергетических изменений. Спектр
сложный и называется
электронно-колебательно-вращательным.
Спектр состоит из полос поглощения.
Максимум полосы поглощения отвечает
наиболее вероятному переходу в данной
области длин волн.
ри
этом имеет место совокупность всех
видов энергетических изменений. Спектр
сложный и называется
электронно-колебательно-вращательным.
Спектр состоит из полос поглощения.
Максимум полосы поглощения отвечает
наиболее вероятному переходу в данной
области длин волн.
На рисунке 2.25 показано относительное расположение энергетических уровней молекулярных орбиталей МО ( и – связывающие МО, * и * – разрыхляющие МО)
В основном состоянии - и -орбитали обычно заняты электронами (это более устойчивое энергетическое состояние с меньшей потенциальной энергией).
Наибольшей энергии требует переход * – проявляется в дальней УФ-области и характерен для молекул насыщенных углеводородов. Переходы * соответствуют видимой и ближней УФ-областям и типичны для молекул ненасыщенных соединений.

Рис.
2.26. Кривые потенциальной энергии
взаимодействия при электронных переходах
![]()
При поглощении крупных квантов лучистой энергии может произойти электронный перескок. Потенциальная энергия диссоциации D0 – уменьшается, а Е – увеличивается. При повышении энергии Е увеличивается межатомное расстояние re в результате колебательного движения (рис 2.26).
Для каждого вида связи имеется своя энергия электронных переходов и своя характерная полоса поглощения с определенной длиной волны.