

Соответственно, если взять производную от отношения по температуре, то уравнение (8.70) предстает в виде:
 .
(10.72)
.
(10.72)
Уравнения (10.69-10.72) известны под названием (соотношений) Гиббса-Гельмгольца. Эти уравнения связывают внутреннюю энергию U с энергией Гельмгольца F, а также энтальпию Н с энергией Гиббса G. Дифференциальные формы уравнений (10.71) и (10.72) являются термодинамически строгими и распространяются на любые термодинамические системы.
Следствие шестое. Уравнение Гиббса-Дюгема. С помощью уравнения, вытекающего из анализа характеристической функции Гиббса можно рассчитать значение химического потенциала одного из компонентов в многокомпонентной системе, если известны значения химических потенциалов других компонентов.
Приведем вывод этого уравнения. При этом будем исходить из ранее полученных уравнений (10.50) и (10.51).
![]()
dG
= -SdТ + Vdр +
![]() .
.
При постоянных т и р уравнение (10.50) можно представить в виде
dGТ,р
=
![]() ,
(10.73)
,
(10.73)
а его полный дифференциал в виде
dGТ,р
=
![]() +
+![]() .
(10.74)
.
(10.74)
При сопоставлении (10.73) с (10.74) получаем
![]() .
(10.75)
.
(10.75)
Уравнение (10.75) получило название уравнения Гиббса-Дюгема. Далее оно будет широко использоваться при термодинамическом описании свойств многокомпонентных систем.
Следствие седьмое. Из всех характеристических функций только внутренняя энергия U обладает свойством сохраняемости; все остальные (Н, F, G) функции даже в изолированной системе могут изменяться.
Запишем фундаментальное уравнение Гиббса для открытой системы (5.11), включив в него все возможные внешние и внутренние взаимодействия. Тогда
![]() (10.76)
(10.76)
Наложим на открытую систему условия изоляции. Тогда в соответствии с изложенным ранее, для полностью изолированной системы dеS=0; dеV=0; dеnк=0 (к=1, …, к). Кроме того, в соотвествии с уравнением де Донде (6.1) для изолированной системы
![]() .
.
Сопоставляя приведенные записи с (10.76) приходим к сделанному ранее выводу (5.4):
dUизол = 0; Uизол = const,
т.е. внутренняя энергия может меняться только за счет внешних взаимодействий.
Так же подробно запишем дифференциальное уравнение для характеристической функции Н, исходя из (10.37):
![]()
(10.77)
При наложении условий полной изоляции (dеS=0;dеV=0;dеnк=0) и учитывая соотношение (6.1), для изменения энтальпии изолированной системы Низолполучим:
dНизол = Vdр. (10.78)
Следовательно, энтальпия системы с химическим превращением не обладает сохраняемостью: она меняется с изменением в изолированной системе давления.
Для характеристической функции F можно, исходя из записи ее полного дифференциала (10.44) записать выражение, применимое для закрытой системы, по аналогии с (10.76) и (10.77):
![]() .
.
(10.79)
С переходом к изолированной системе, полностью лишенной возможности взаимодействия с окружающей средой, в уравнении (10.79) исчезают лишь второе и третье слагаемое:
![]() .
(10.80)
.
(10.80)
Следовательно, энергия Гельмгольца изолированной системы с химическим превращением способна меняться за счет изменения температуры и числа молей компонентов химической реакции.
Исходя из выражения для полного дифференциала энергии Гиббса G (8.51) для открытой системы и переходя затем к изолированной системе за счет наложения на нее изоляций, для полностью изолированной системы получаем:
![]() .
(10.81)
.
(10.81)
Следовательно, энергия Гиббса в случае изолированной системы с химическим превращением полностью сохраняет возможность меняться в результате изменения всех ее характеристических аргументов – температуры, давления и числа молей веществ – участников химической реакции.
Таким образом, наряду с общими признаками, характеристические функции имеют отличия, связанные с их сохраняемостью.
10.3. Характеристические функции в роли
термодинамических потенциалов
В этом разделе дается изложение еще одного следствия, помимо описанных выше, вытекающего из анализа характеристических функций.
Суть его состоит в том, что характеристические функции при определенных условиях могут выступать в роли термодинамических потенциалов. Термодинамическим потенциалом называют характеристическую функцию, убыль которой в обратимом процессе, протекающем при постоянстве собственных переменных, равна максимально полезной работе, а в необратимом процессе – некомпенсированной теплоте.
Наиболее общим соотношением для бесконечно малого необратимого изменения в закрытой системе является обобщенное уравнение первого и второго начал термодинамики (7.1)
dU = ТdS – рdV – dQ’ ,
где Q’ – избыточная некомпенсированная теплота, выделяющаяся в результате внутренних необратимых процессов в системе (dQ’ > 0).
Такими процессами, как уже указывалось, могут быть химические, электрические, электрохимические, фазовые превращения и другие. Они в данном уравнении учитываются суммарно в виде dQ’ , как превращающиеся в некомпенсированную теплоту. Если процессы, происходящие внутри системы, стараются учесть, оценивая конкретные работы или определяя суммарную работу А, то ее записывают в уравнение (7.1) вместо некомпенсированной теплоты:
dU = ТdS – рdV – А’maх , (10.82)
где А’maх – максимальная полезная работа немеханической природы (полезная работа в обратимом процессе).
Уравнение (10.82), наряду с (7.1) также называют объединенным уравнением первого и второго начал термодинамики. Оно является одним из основных фундаментальных уравнений в химической термодинамике.
Если системой производится только работа расширения (сжатия), то объединенное уравнение (10.82) принимает известный вид (5.5) для обратимых процессов:
dU = ТdS – рdV.
Для необратимых термомеханических процессов справедливо неравенство:
ТdS > dU + рdV. (10.83)
Существование этого неравенства связано с тем, что как указывалось ранее (раздел 6.1) работа обратимого (равновесного) процесса имеет максимальную величину по сравнению с работой необратимого (неравновесного) процесса. По этой причине работу обратимого процесса называют максимальной работой Аmaх.
Очевидно, что для термодинамических расчетов обычно используются уравнения, описывающие обратимые равновесные процессы, типа (5.5) и (10.82), поскольку они позволяют вычислять не только свойства систем, но и функции процессов, которые в определенных условиях приобретают свойства функций состояния и перестают зависеть от пути процесса.
Другими словами, при условии постоянства естественных переменных максимальная полезная работа немеханической природы А’maх в уравнении (10.82) приобретает свойства функции состояния, а ее значение определяется изменением соответствующего термодинамического потенциала (разностью потенциалов), в роли которых выступают соответствующие характеристические функции (U, Н, F, G).
В соответствии с (10.82) дифференциалы четырех характеристических функций (U, Н, F, G) можно выразить в виде соотношений, включающих максимально полезную работу немеханической природы:
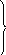 dU = ТdS – рdV – А’maх
,
dU = ТdS – рdV – А’maх
,
dН = ТdS – Vdр – А’maх ,
dF = - SdТ – рdV – А’maх , (10.84)
dG = -SdТ – Vdр – А’maх ,
Исходя из этих выражений определим следующие термодинамические потенциалы:
при S=Const и V=Const
dU = – А’maх или -U = А’maх , (10.85)
т.е. внутренняя энергия является изохорно-изоэнтропийным потенциалом;
при S=Const и р=Const
dН = – А’maх или -Н = А’maх, (10.86)
т.е. энтальпия является изобарно-изоэнтропийным потенциалом;
3. при Т=Const и V=Const
dF = – А’maх или -F = А’maх , (10.87)
т.е. энергия Гельмгольца является изохорно-изотермическим потенциалом;
при Т=Const и р=Const
dG = – А’maх или -G = А’maх , (10.88)
т.е. энергия Гиббса является изобарно-изотермическим потенциалом.
Таким образом, убыль любого термодинамического потенциала в обратимом процессе при постоянных значениях соответствующих ему естественных переменных равна максимально полезной работе немеханической природы. Роль термодинамических потенциалов аналогична роли потенциальной энергии, определяющей максимальное количество механической работы при перемещении тела в поле сил тяготения.
10.4. Критерии самопроизвольного протекания процессов
Как показано в разделе 6.2, направленность самопроизвольных процессов в изолированных системах определяется знаком изменения энтропии. Поскольку все самопроизвольные процессы являются необратимыми, то для изолированной системы критерием самопроизвольного протекания процессов служит положительное изменение (рост) энтропии (6.6):
(diS)изол
![]() 0.
0.
По мере приближения системы к равновесному состоянию рост энтропии снижается и она достигает максимального значения в состоянии равновесия. При этом (diSизол = 0) – знак равенства относится к стационарному равновесному состоянию.
Направление самопроизвольных (необратимых) процессов, протекающих в более сложных (закрытых и открытых) системах определяют по изменению термодинамических потенциалов. Поскольку в необратимом процессе количество работы, которое может быть получено, оказывается меньше максимально возможной величины работы в обратимом процессе, возникают неравенства:
 А’
-dUS,V
; А’
-dНS,Р
А’
-dUS,V
; А’
-dНS,Р
А’ -dFV,Т ; А’ -dGР,Т (10.89)
Все неравенства (10.89) выведены из условия А’ А’maх , т.е. неравенства полезной работы А’ в необратимом процессе и максимально полезной работы А’maх в обратимом процессе.
Поскольку полезная работа всегда положительна, то очевидно, что изменения всех потенциалов при постоянстве их естественных переменных в самопроизвольном (необратимом) процессе всегда отрицательны:
dUS,V 0; dНS,Р 0; dFV,Т 0; dGР,Т 0. (10.90)
Другими словами, самопроизвольный процесс в закрытой или открытой системе сопровождается уменьшением термодинамических потенциалов при условии постоянства соответствующих им естественных параметров.
Любой самопроизвольный процесс стремится к равновесию, а уменьшающиеся термодинамические потенциалы, очевидно, при установлении равновесия в системе становятся исчерпанными и достигают минимальных величин при соответствующих постоянных параметрах. Поэтому критериями равновесия в системе являются соотношения:
dUS,V = 0; dНS,Р = 0; dFV,Т = 0; dGР,Т = 0. (10.91)
В качестве критериев направленности процессов и равновесия в системе обычно используют энергию Гиббса или энергию Гельмгольца, так как реальные процессы чаще всего происходят при постоянной температуре и давлении (или объеме). Условия постоянства энтропии и контроль за ее изменением осуществить практически невозможно, что исключает использование внутренней энергии, энтальпии и энтропии для практической оценки направленности процессов и достижения равновесия.
Вместе с тем имеется возможность перехода от одних термодинамических потенциалов к другим, благодаря соотношениям Гиббса-Гельмгольца (10.69) и (10.70), полученным в виде одного из следствий анализа характеристических функций. Так, соотношения (10.69) и (10.70) дают возможность установить связь между максимальной работой процесса А’maх , протекающего равновесно, и теплотой того же процесса, но протекающего неравновесно.
В качестве примера рассмотрим обратимый изотермический переход системы из состояния 1 в состояние 2 при постоянном давлении (dТ=0; dр=0). Согласно уравнению Гиббса-Гельмгольца (10.69) можно записать для закрытой системы:
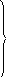
![]()
![]() .
(10.92)
.
(10.92)
Вычитая одно выражение из другого, получим
![]() .
(10.93)
.
(10.93)
На основании (10.88)
-(G)Т,Р = А’maх
имеем
![]() .
(10.94)
.
(10.94)
С другой стороны, убыль энтропии системы при постоянном давлении равна количеству выделившейся теплоты в ходе химической реакции (7.8):
QР = -НР .
С учетом уравнения (7.8) уравнение (10.94) приобретает вид:
![]() .
(10.95)
.
(10.95)
В
уравнении (10.95)
QР
является тепловым эффектом химической
реакции, протекающей необратимо.
Следовательно, величина QР
может быть определена термохимическим
способом (калориметрически или путем
вычисления на основании закона Гесса).
Произведение
![]() ,
равное ТS,
представляет собой теплоту, которая
отдается системой внешней среде или,
наоборот, поглощается из нее и тем самым
поддерживает изотермические условия
в системе. Таким образом, система должна
находится в тепловом контакте с внешней
средой, температура которой постоянна
и равна температуре в системе. Такой
обмен теплотой между системой и окружающей
средой можно считать обратимым процессом.
,
равное ТS,
представляет собой теплоту, которая
отдается системой внешней среде или,
наоборот, поглощается из нее и тем самым
поддерживает изотермические условия
в системе. Таким образом, система должна
находится в тепловом контакте с внешней
средой, температура которой постоянна
и равна температуре в системе. Такой
обмен теплотой между системой и окружающей
средой можно считать обратимым процессом.
Исходя из изложенного уравнение (10.95) можно записать в виде:
![]() .
(10.96)
.
(10.96)
Аналогичным образом получается уравнение для энергии Гельмгольца:
![]() .
(10.97)
.
(10.97)
Из уравнений (10.96) и (10.97) видно, что даже совершая максимально полезную работу, система часть своей энергии может терять в виде теплоты (при протекании экзотермических реакций, когда QР 0; QV 0, или что то же самое, Н 0; U 0.) К этому следует добавить, что еще более интересная ситуация возникает при эндотермических реакциях, когда QР0 или QV 0, или, что то же самое, Н 0; U 0. В этом случае единственной движущей силой самопроизвольно протекающего процесса является увеличение теплоты системы, а источником этой теплоты может быть только окружающая среда. Таким образом, мы имеем несколько критериев самопроизвольного протекания процессов.
Т а б л и ц а 10.1
Критерии самопроизвольности процессов
|
Функции |
S |
F |
G |
|
Определение |
|
F = U - ТS |
G = Н – ТS |
|
Измерение функций |
|
|
|
|
Ограничения |
Изолированная система |
Т = Const V = Const |
Т = Const Р = Const
|
|
Критерии самопроизвольности процессов |
S 0 |
F 0 |
G 0 |
|
Условия равновесия
|
Максимум S dS = 0 |
Минимум F dF = 0 |
Минимум G dG = 0 |
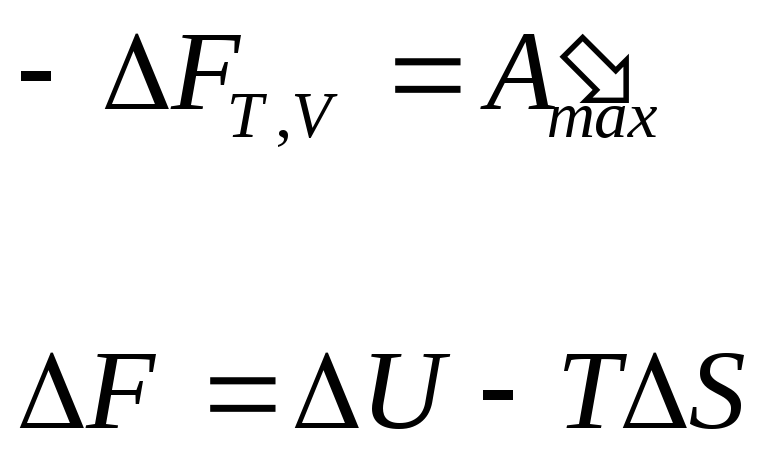
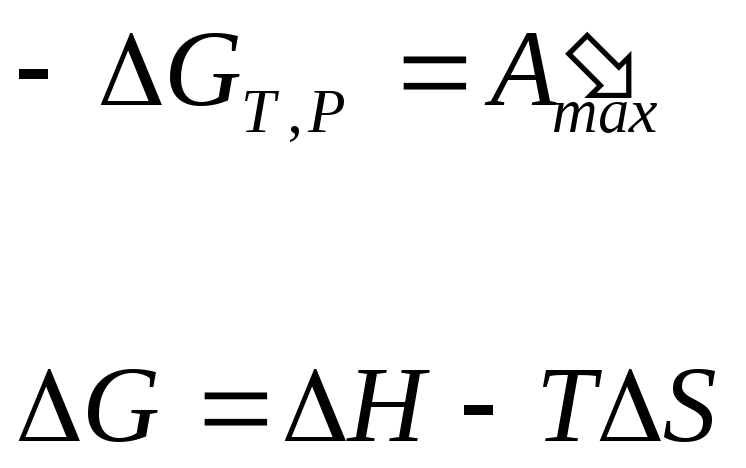
Из таблицы следует, что для самопроизвольно протекающих процессов существуют две движущие силы: стремление перейти в состояние с наименьшей энергией (выделить тепло) и стремление перейти в наиболее вероятное энергетическое состояние (с максимальным числом микросостояний системы – с максимумом энтропии).
При протекании физико-химических процессов в закрытых или открытых системах в общем случае одновременно изменяются и энергия системы и энтропия. Процесс идет в направлении, при котором общая (суммарная) движущая сила процесса (ее термодинамический потенциал) уменьшается: F 0; G 0.
10.5. Стандартные термодинамические потенциалы
Как было показано ранее, характеристические функции являются функциями состояния системы и зависят только от начального и конечного состояния системы. Для оценки и сопоставления термодинамических потенциалов, в роли которых выступают характеристические функции, при проведении химических реакций в различных условиях вводится понятие стандартных термодинамических потенциалов G0 и F0.
По
аналогии с тепловыми эффектами реакций
(закон Гесса) стандартные значения G0
и F0
могут быть найдены из стандартных
значений
![]() и
и![]() образования веществ, принимающих участие
в реакции:
образования веществ, принимающих участие
в реакции:
![]()
![]() (10.98)
(10.98)
С учетом знаков стехиометрических коэффициентов
![]()
![]() (10.99)
(10.99)
Поскольку
в большинстве случаев химические
процессы удобнее изучать при постоянных
Т и Р, наибольший практический интерес
вызывает анализ изменения термодинамического
потенциала G.
Для расчетов G0
удобно пользоваться таблицами стандартных
энергий Гиббса образования соединений
![]() ,
подобно таблицам тепловых эффектов
образования
,
подобно таблицам тепловых эффектов
образования![]() и энтропии образования
и энтропии образования![]() соединений.
соединений.
Тот факт, что абсолютное значение энергии Гиббса не может быть определено, не имеет существенного значения, так как для решения термодинамических задач требуется определять изменение этой величины. Это допускает условность в выборе начала отсчета энергий Гиббса.
Принято считать, что энергии Гиббса образования всех элементов в стандартном состоянии равны нулю. Стандартными состояниями твердых и жидких веществ, как указывалось ранее, являются их стабильные формы при стандартных параметрах. Отсюда стандартные энергии Гиббса соединений равны изменению энергий Гиббса их образования, т.е. равны изменению энергии Гиббса, сопровождающему образование 1 моля соединения из элементов, находящихся в стандартных состояниях (стандартные энергии Гиббса образования соединений обычно приводятся в таблицах при Т=298К и Р=1атм.
Таким
образом, стандартным
изобарно-изотермическим потенциалом
образования
![]() называется изменение энергии Гиббса
для реакции, по которой 1 моль вещества
в стандартном состоянии образуется из
простых веществ в их стандартных
состояниях.
называется изменение энергии Гиббса
для реакции, по которой 1 моль вещества
в стандартном состоянии образуется из
простых веществ в их стандартных
состояниях.
Например, для реакции между водородом и кислородом при стандартных условиях
Н2(г)
+ ½ О2(г)
= Н2О(г)
;
![]() =-229,0
кДж/кмоль.
=-229,0
кДж/кмоль.
Это значит, что энергия Гиббса 1 моля водяного пара в стандартном состоянии на 229 кДж меньше, чем сумма энергий Гиббса 1 моля газообразного водорода и ½ моля газообразного кислорода в их стандартных состояниях при Т=298К:
![]() .
.
Так как стандартные энергии Гиббса элементов (водорода и кислорода) приняты равными нулю, то стандартная энергия 1 моля водяного пара становится равной стандартной энергии Гиббса его образования из составляющих элементов.
Для
многих химических реакций в таблицах
стандартных величин отсутсвуют значения
![]() ,
а содержатся только значения
,
а содержатся только значения![]() и
и![]() .
.
В таких случаях, используя выражение (10.49)
![]() ,
,
справедливое для процесса при Т=Сonst, можно стандартную энергию Гиббса выразить в виде
![]() , (10.100)
, (10.100)
где
![]() и
и![]() определяются соответственно выражениями
определяются соответственно выражениями
![]()
![]() ,
(10.101)
,
(10.101)
где «к» и «н» относятся соответственно к конечному и начальному состояниям.
Все
расчеты с помощью таблиц стандартных
величин основываются на аддитивном
свойстве величин
![]() ,
,![]() и
и![]() и поэтому сводятся к простому
алгебраическому суммированию по аналогии
с расчетами, основанными на законе
Гесса.
и поэтому сводятся к простому
алгебраическому суммированию по аналогии
с расчетами, основанными на законе
Гесса.
Пример:
Рассчитать стандартную энергию Гиббса реакции
CH4(г) + 4 SO2Cl2 = CCl4(ж) + 4 SO2(г) 4 HCl(г)
по стандартным величинам энтальпии и энтропии
Решение:
Выпишем из справочника необходимые сведения об участниках реакции.
|
|
CH4(г) |
SO2Cl2(ж) |
CCl4(ж) |
SO2(г) |
HCl(г) |
|
|
–50,85 |
–321,49 |
–62,66 |
–300,21 |
–95,3 |
|
|
–74,85 |
–394,13 |
–132,84 |
–296,9 |
–92,31 |
|
|
186,27 |
216,31 |
216,19 |
248,07 |
186,79 |
![]()
![]() =
–62,66 + 4(–300,21) + 4 (–95,3) – (–50,85) – 4 (–321,49)
= – 307,85 кДж/моль.
=
–62,66 + 4(–300,21) + 4 (–95,3) – (–50,85) – 4 (–321,49)
= – 307,85 кДж/моль.
Энергию Гиббса можно рассчитать, используя уравнения 8.100 и 8.101.
![]() =
– 132,84 + 4(–296,9) + 4 (–92,31) – (–74,85) – 4
(–394,13) = – 38,31 кДж/моль.
=
– 132,84 + 4(–296,9) + 4 (–92,31) – (–74,85) – 4
(–394,13) = – 38,31 кДж/моль.
![]() =
216,19 + 4 248,07 + 4 186,79 – 186,27 – 4 216,31 = 904,12 Дж/моль
К.
=
216,19 + 4 248,07 + 4 186,79 – 186,27 – 4 216,31 = 904,12 Дж/моль
К.
![]() =
=
![]() =
–38310 – 218 904,12 = – 307737,76 Дж/моль
=
=
–38310 – 218 904,12 = – 307737,76 Дж/моль
=
– 307,74 кДж/моль.
Энергия Гиббса рассчитанная обоими способами практически одинаковая.
Переход
от расчетных значений
![]() к
к![]() при постоянстве температуры достаточно
просто осуществить, благодаря соотношению,
вытекающему из определения функций G и
F:
при постоянстве температуры достаточно
просто осуществить, благодаря соотношению,
вытекающему из определения функций G и
F:
![]() .
(10.102)
.
(10.102)
Для реакций в конденсированных средах
![]() .
(10.103)
.
(10.103)
Если в реакции участвуют газы и можно считать, что они ведут себя как идеальные, то с учетом уравнения Менделеева-Клапейрона
![]() ,
(10.104)
,
(10.104)
где n – изменение числа молей компонентов в ходе реакции.
При n = 0
![]() .
(10.105)
.
(10.105)
Направление изменений энергии Гиббса и энергии Гельмгольца (увеличение или уменьшение) при изменении их естественных переменных по сравнению со стандартными условиями определяется знаком частных производных этих термодинамических потенциалов по этим переменным.
Так, из соотношений (10.24) и (10.25)
![]() ;
;
![]()
следует общий вывод: поскольку значения объема и энтропии при любой температуре (кроме 0 К) и любом давлении всегда положительны, частная производная энергии Гиббса по температуре при Р = Const всегда отрицательна, а по давлению при Т = Const – положительна. Следовательно, энергия Гиббса с повышением температуры при Р = Const убывает, а с возрастанием давления при Т = Const – увеличивается.
Зависимость энергии Гельмгольца от объема системы и температуры раскрывают уравнения (10.17) и (10.18):
![]() ;
;
![]() .
.
Эти соотношения показывают, что энергия Гельмгольца убывает, как с повышением температуры при V = Const, так и с увеличением объема системы при Т = Const.
Полученные количественные выводы, однако, недостаточны для количественного решения термодинасмических задач.
Переход от термодинамических потенциалов при стандартной температуре к потенциалам при любой другой температуре может быть осуществлен двумя способами: с помощью уравнгений Гиббса-Гельмгольца в виде (10.71) или (10.72), а также с помощью привычных для расчетов выражений для энергии Гиббса и Гельмгольца (10.42) и (10.49).
Как тот, так и другой способы определения стандартных потенциалов при любой произвольной температуре требуют использования их зависимостей от температуры для конкретной химической реакции. Полученные в соответствующих главах выражения позволяют определить, например, на основе соотношения (10.49) стандартные энергии Гиббса при любой температуре, отличной от стандартной (298 К). Тем не менее, для облегчения восприятия метода, ниже приведен порядок расчета, необходимого для решения инженерных вопросов с использованием справочных данных.
1. Определение стандартного теплового эффекта реакции (изменение энтальпии системы при химических превращениях в стандартных условиях). При стандартных условиях в соответствии с уравнением (10.101).
![]() .
.
2. Определение стандартного теплового эффекта реакции при произвольной температуре. На основании записи уравнения Кирхгофа (7.38) в виде (7.41)
![]()
и эмпирических зависимостей СР от температуры с использованием температурных коэффициентов а, в, с, с` (5.46)
СР = а + в + сТ2 + с`Т-2
при совместном решении (7.41) и (7.46) получаем стандартный тепловой эффект реакции при произвольной температуре Т (7.49):
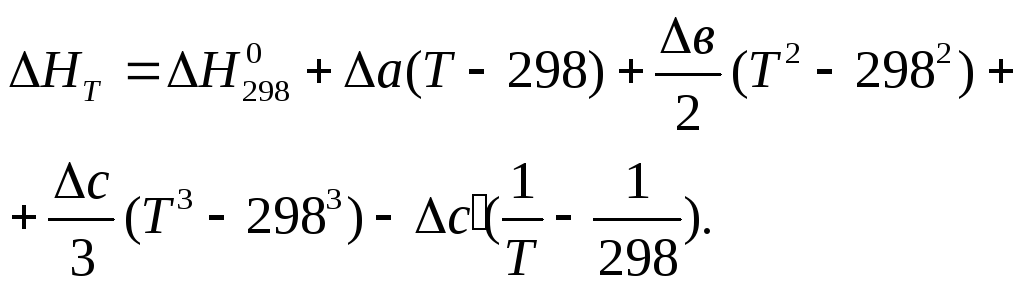
Температурные коэффициенты для каждого из веществ – участников реакции приводятся, наряду с другими характеристиками веществ, (Н0, S0) в справочниках.
3. Определение стандартного изменения энтропии реакции. При стандартных условиях
![]() .
.
4. Определение стандартного изменения энтропии реакции при произвольной температуре. На основании записи (6.32)
![]()
и эмпирических зависимостей СР от температуры типа (7.46) при их совместном решении получаем стандартные изменения энтропии реакции при произвольной температуре (8.33) в виде
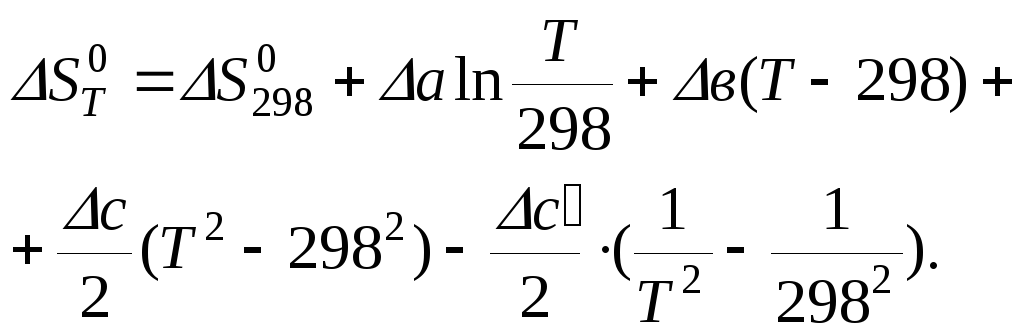
5. Определение стандартного термодинамического потенциала Гиббса химической реакции. При стандартных условиях в соответствии с (10.98)
![]()
и (10.100)
![]() .
.
Подставляя
соответствующие значения
![]() и
и![]() ,
вычисленные ранее, в последнее уравнение,
получаем
,
вычисленные ранее, в последнее уравнение,
получаем![]() .
.
6.
Определение стандартного изменения
энергии Гиббса химической реакции при
произвольной температуре. При температуре
Т
![]() .
Подставляя соответствующие выражения
для
.
Подставляя соответствующие выражения
для![]() и для
и для![]() ,
это уравнение можно записать в виде:
,
это уравнение можно записать в виде:
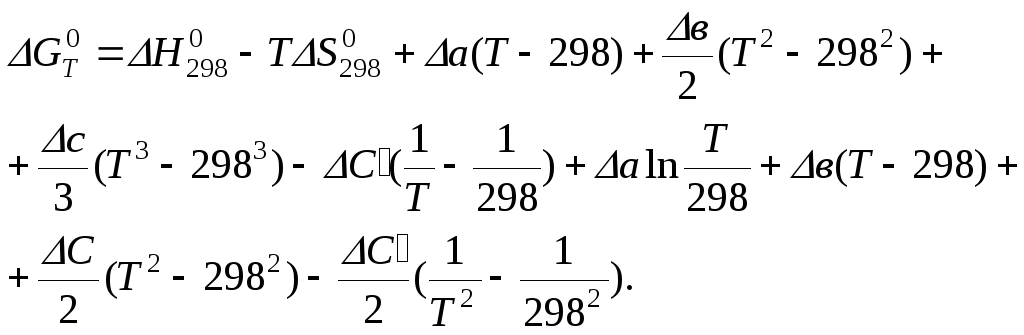
Следует заметить, что температурные коэффициенты а, в, с, с` для каждого из веществ приводятся в справочных таблицах для вполне определенного интервала температур, ограниченного устойчивым агрегатным состоянием веществ.
7.
Результатом проведенных расчетов
является определение направления
самопроизвольного химического процесса.
Если расчетное значение
![]() ,
то процесс при стандартных условиях
протекает самопроизвольно; если
,
то процесс при стандартных условиях
протекает самопроизвольно; если![]() ,
то для проведения процесса требуется
подведение дополнительной энергии к
системе извне.
,
то для проведения процесса требуется
подведение дополнительной энергии к
системе извне.
Пример.
Рассчитать энергию Гиббса реакции
CH4(г) + 4SO2Cl2(г) = CCl4(г) + 4SO2(г) + 4HCl(г)
при температуре 1000 К и давлении 1,013 105 Па
Решение
Значения ΔНо298, Δа, Δb, Δс, Δс/ возьмем из задачи раздела 7.6.
ΔНо298 = -129,73кДж/моль,
Δа = -15,72; Δb = 7,62 10-3; Δс = 20,15 10-6; Δс/ = 30,48 105.
ΔSо298 рассчитаем по справочным данным
|
|
CH4(г) |
SO2Cl2(г) |
CCl4(г) |
SO2(г) |
HCl(г) |
|
|
186,27 |
311,29 |
310,12 |
248,07 |
186,79 |
![]() =
310,12 + 4(248,07 + 4 186,79 – 186,27 – 4 311,29) = 618,13
Дж/моль
К
=
310,12 + 4(248,07 + 4 186,79 – 186,27 – 4 311,29) = 618,13
Дж/моль
К
![]() =
=![]()
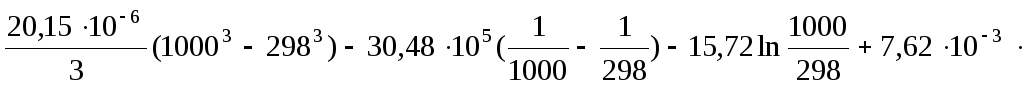
![]()
=-741693,541 Дж/моль.
Следует также предостеречь от ошибочного вывода (особенно относительно реакций в газовой среде), заключающегося в том, что положительная величина G0 означает полную невозможность протекания данной реакции в прямом направлении и получения технологически приемлемого выхода ее продуктов. Величина G0 характеризует направленность процесса для газовых реакций лишь для стандартного состояния, когда все участники реакций имеют одинаковые парциальные давления, равные атмосферному. Если же изменить эти давления, то может поменяться и направление реакции.
Кроме того, направленность протекания газовых реакций в сильной степени зависит и от общего давления. Зависимость энергии Гиббса от давления следует, например, из соотношения (10.21) при условии постоянства температуры:
![]() ;
;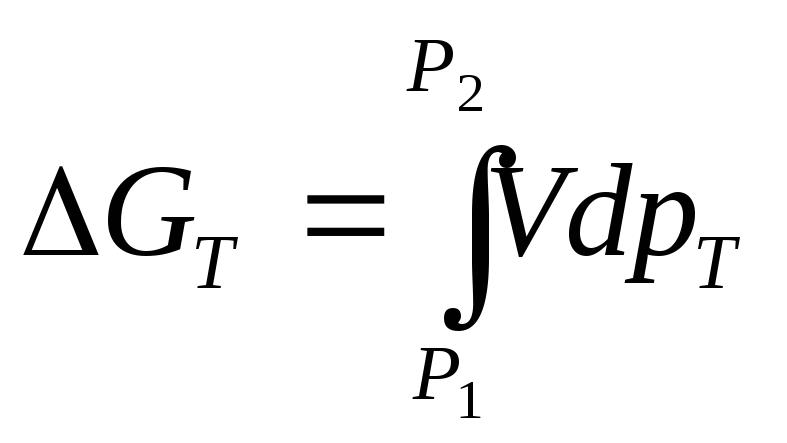 .
(10.106)
.
(10.106)
Чтобы вычислить интеграл, надо знать зависимость объема от давления при постоянной температуре, т.е. уравнение состояния. Предположив, что система является идеальным газом, для которого может быть использовано известное уравнение состояния (5.18), найдем решение уравнения (10.106) для одного моля газа:
 .
(10.107)
.
(10.107)
Если известна энергия Гиббса G1 при некотором давлении р1, то получим следующую зависимость:
![]() .
(10.108)
.
(10.108)
Отсюда следует, что энергия Гиббса идеального газа при постоянной температуре возрастает пропорционально логарифму давления газа.
Следует
отметить, что термодинамические
потенциалы дают, в основном, информацию
о направленности процесса, но не содержат
информации о его интенсивности. Например,
для реакции образования СО2
из графита и кислорода
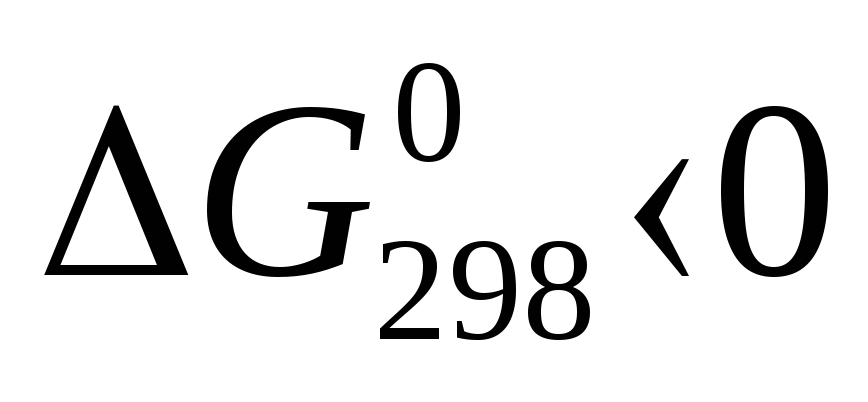 ,
но графит с кислородом практически не
реагируют при 298 К. Чтобы такой процесс
пошел, необходимо создать определенные
условия для увеличения скорости процесса.
Таким образом, наряду с оценкой
принципиальной возможности протекания
процесса, должен существовать критерий
(или критерии), с помощью которого
(которых) можно оценивать и регулировать
на практике интенсивность протекания
процессов.
,
но графит с кислородом практически не
реагируют при 298 К. Чтобы такой процесс
пошел, необходимо создать определенные
условия для увеличения скорости процесса.
Таким образом, наряду с оценкой
принципиальной возможности протекания
процесса, должен существовать критерий
(или критерии), с помощью которого
(которых) можно оценивать и регулировать
на практике интенсивность протекания
процессов.
10.6. Мера химического сродства
Способность различных веществ вступать в химическое взаимодействие с образованием новых, отличающихся по своим свойствам веществ получила название химического сродства. Этот термин не совсем удачен, так как не отражает действительного смысла обозначаемого им понятия, но он сохранился до настоящего времени. В действительности под химическим сродством понимают совокупность химических и физических условий, которая в данной системе создает возможность протекания физико-химического процесса, связанного с изменением состава системы.
Чтобы связать понятие сродства с какой либо количественной характеристикой, определяющей одновременно интенсивность и направленность физико-химического процесса, нужен критерий, который мог бы служить мерой сродства. Он был найден далеко не сразу.
Предлагалось выражать меру химического сродства скоростью химической реакции. Однако это предложение было со временем отвергнуто, потому что сродство химических реакций зависит не только от параметров состояния реагирующих веществ (температура, давление, концентрация), но и от способа проведения реакции (стационарный или нестационарный режимы, способы смешения реагентов и вывода продуктов из зоны реакции, отсутствие или присутствие катализатора и др.).
Предложение принять в качестве меры химического сродства положительный тепловой эффект реакции (Бертло, 1867г.) также оказалось неудачным, так как наряду с экзотермическими реакциями известно много самопроизвольно и энергично протекающих эндотермических реакций.
Гиббс (1878г.) и Гельмгольц (1884г.) предложили оценивать химическое сродство значением максимально полезной работы, которую силы, заставляющие вещества взаимодействовать, способны произвести. Позже Вант-Гофф (1885г.) пришел независимо к такому же выводу.
В процессах при Т,V=Сonst и Т,Р=Const, т.е. в условиях, при которых обычно рассматриваются химические реакции, максимальное количество немеханической работы равно (см. раздел 10.3) соответственно уменьшению энергии Гельмгольца (-F) и энергии Гиббса (-G). Следовательно, в сложной системе при рассматриваемых условиях могут протекать только такие химические реакции, которые ведут к уменьшению абсолютных значений термодинамических потенциалов, а значения этого уменьшения при переходе системы из неравновесного состояния в равновесное являются мерой химического сродства реагирующих веществ.
Таким образом, термодинамические потенциалы G и F удовлетворяют всем требованиям, которые могут быть предъявлены к величинам, характеризующим химическое сродство:
G и F не зависят от пути, по которому протекает реакция, а зависят от свойств компонентов и их состояния;
знак G или F определяет направление процесса;
при равновесии G или F минимальны, т.е. самопроизвольные процессы в системе исключаются.
Вместе с тем, как уже указывалось ранее (см. раздел 10.5) при использовании термодинамических потенциалов (в частности G) часто возникает несоответствие между оценкой принципиальной возможности протекания химического процесса и его реальным выражением (интенсивностью химических превращений).
Ограниченность метода Гиббса-Гельмгольца заключается в том, что применение величин, относящихся непосредственно к химической реакции (максимальная работа), приводит к неудобствам, связанным с тем, что указанные величины не являются функциями состояния системы в каждый данный момент времени, но зависят от условий течения реакции (р=Сonst или V= Сonst), что приводит к неопределенностям. Методу Гиббса-Гельмгольца, таким образом, недостает функции состояния, связанной непосредственно с химической реакцией. Кроме того, в этом методе, по существу, рассматриваются только равновесные состояния и обратимые изменения, хотя, как было показано, такая величина, как теплота реакции, имеет ясный смысл лишь в том случае, когда химическая реакция протекает на определенную глубину за конечное время. Следовательно, любую химическую реакцию (см. раздел 6.3) следует рассматривать как необратимое явление.
В качестве примера рассмотрим обратимую диссоциацию газа. Водяной пар в закрытом сосуде при 12000С диссоциирует до установления равновесия
Н2О Н2 + ½ О2.
При
этой температуре и заданном давлении
мольная доля воды в смеси газов
![]() имеет вполне определенное значение.
Этому равновесному состоянию отвечает
точка А на рис. 10.1.
имеет вполне определенное значение.
Этому равновесному состоянию отвечает
точка А на рис. 10.1.
Если
немного повысить температуру и перейти
в точку А/,
то системы выйдет из равновесного
состояния. При этом количество молекул
воды, подвергшихся диссоциации, увеличится
(уменьшится
![]() ),
и система будет стремиться перейти в
равновесное состояние, соответствующее
точке А*
на линии равновесия АВ. Если, однако,
температура постепенно повышается, то
изменение состояния системы описывается
линией А`В`, которая при соответствующем
выборе скорости изменения температуры
будет очень близка к АВ. В точке В/
прекратим повышение температуры и
позволим перейти системе в равновесное
состояние В. Из этой точки можно вернуть
систему в точку А, понижая температуру
и и проводя систему черезпоследовательность
состояний, соответствующих линии
),
и система будет стремиться перейти в
равновесное состояние, соответствующее
точке А*
на линии равновесия АВ. Если, однако,
температура постепенно повышается, то
изменение состояния системы описывается
линией А`В`, которая при соответствующем
выборе скорости изменения температуры
будет очень близка к АВ. В точке В/
прекратим повышение температуры и
позволим перейти системе в равновесное
состояние В. Из этой точки можно вернуть
систему в точку А, понижая температуру
и и проводя систему черезпоследовательность
состояний, соответствующих линии
![]()
![]() .Из
рисунка видно, что на пути
.Из
рисунка видно, что на пути![]()
![]() система не проходит через те же состояния,
что на пути
система не проходит через те же состояния,
что на пути![]() ,
поэтому реальный процесс диссоциации
паров воды необратим. Однако при
соответствующем контроле за изменениями
температуры всегда можно поддерживать
температуру сколь угодно близкой к
равновесной, в то же время не позволяя
ей принять точное равновесное значение,
так как иначе реакция прекратится. Таким
способом можно сде лать процесс
,
поэтому реальный процесс диссоциации
паров воды необратим. Однако при
соответствующем контроле за изменениями
температуры всегда можно поддерживать
температуру сколь угодно близкой к
равновесной, в то же время не позволяя
ей принять точное равновесное значение,
так как иначе реакция прекратится. Таким
способом можно сде лать процесс![]() сколь угодно близким к процессу АВ,
рассматривая АВ как идеализированный
процесс реального процесса
сколь угодно близким к процессу АВ,
рассматривая АВ как идеализированный
процесс реального процесса![]() .
.

 D
D
Т
Рис. 10.1. Обратимая диссоциация воды.
Как видно из приведенного примера, все реальные явления необратимы. Об обратимых процессах можно говорить только как о гипотетических идеализированных процессах. Существует, однако, множество явлений, которые нельзя идеализировать подобным образом. Рассмотрим, например, систему в состоянии, соответствующем точке «С» на рис. 10.1, и предположим, что температура системы поддерживается постоянной (Т=Const). Тогда система будет проходить через состояния, изображаемые точками отрезка СD. Этот процесс нельзя провести в обратном порядке вдоль DС, так как в каждой точке этого отрезка скорость диссоциации молекул воды больше скорости их образования. Таким образом, изменение СD не может быть связано с каким либо идеальным обратимым процессом.
Важнейшим достижением термодинамики за последнее время является ее распространение на процессы, необратимые по своей природе. В серии работ, первая из которых появилась еще в 1920г., де Донде развил новые представления в химической термодинамике, детально разработанные в дальнейшем И. Пригожиным и Р. Дэфэем.
Рассмотрим основные положения этого метода. С уравнением де Донде, описывающим самопроизвольно протекающие необратимые химические процессы, мы уже встречались (см. раздел 6.2) в виде (6.2)
![]() ;
;
и в виде (6.3)
![]() ,
,
где
![]() - теплота необратимого самопроизвольного
процесса (или некомпенсированная
теплота).
- теплота необратимого самопроизвольного
процесса (или некомпенсированная
теплота).
Из этих уравнений непосредственно вытекает математическое выражение второго начала термодинамики (6.6)
![]() ,
,
согласно которому изменение энтропии для необратимых процессов внутри системы всегда положительно, а для обратимых изменений внутри системы равно нулю.
Главная
идея подхода де Донде заключается в
том, что при рассмотрении химической
реакции, как необратимого процесса,
можно пойти дальше неравенства второго
начала термодинамики и дать количественное
определение некомпенсированной теплоты
![]() (или возрастания энтропии diS)
непосредственно в ходе течения реакции.
(или возрастания энтропии diS)
непосредственно в ходе течения реакции.
При рассмотрении функций состояния и функций процессов по отношению к системам, в которых протекает химическая реакция, в качестве полезной работы принимают химическую работу, в результате которой происходит перераспределение масс веществ в системе, т.е. изменяется состав системы. Системы, которые при этом рассматриваются, находятся в состоянии частичного равновесия. Это значит, что по отношению к некоторым переменным (температуре и давлению) равновесие уже установилось, и необратимость не связана с изменениями этих переменных. В то же время равновесие еще не достигнуто по отношению к распределению вещества между компонентами, способными к химическому взаимодействию, по отношению к распределению вещества между различными фазами системы или подсистемами и в общем случае к любым изменениям, которые можно охарактеризовать параметром (химической переменной) – см. раздел 3.4.
Простым примером систем такого рода является смесь идеальных газов, способных к химическому взаимодействию, в которой имеет место максвелловское распределение молекул по скоростям, но концентрации не соответствуют химическому равновесию между компонентами. В такой системе возможно одно изменение, характеризуемое в любой момент времени величиной химической переменной .
Пусть за время dt значение изменилось на величину d. Так как рассматриваемое изменение является единственным необратимым процессом в системе, то возрастание энтропии должно определяться только величиной d. Поэтому можно записать
![]() .
(10.109)
.
(10.109)
Неравенство соответствует самопроизвольному протеканию реакции, равенство – равновесию. Этим фундаментальным соотношением, впервые сформулированным де Донде, вводится функция состояния А*, названная химическим сродством реакции. По сути химическое сродство А* является функцией мгновенного состояния системы при течении реакции. В новом изложении это характеристика способности веществ к их превращению, она отражает движущую силу перераспределения компонентов как за счет химической реакции, так и вследствие фазового перехода или переноса вещества из одной части системы в другую. Запас химической энергии и ее изменение, т.е. совершаемая химическая работа, представляет собой произведение фактора интенсивности А* (обобщенного потенциала) на фактор емкости (обобщенной координаты), а приращение химической энергии есть произведение А*d.
При этом определении химического сродства А* нет необходимости ограничивать дальнейшее рассмотрение химического сродства состоянием частичного равновесия. Отметим также, что неравенство (10.109) применимо также и к открытым системам. Открытые системы в отличие от закрытых обмениваются с внешней средой и веществом и энергией. Для открытых систем наряду с потоком энтропии dеS(тепл.) возможен и перенос энтропии, связанный с переносом веществ dеSмасс (5.9). Выражение же для прироста энтропии за счет химических реакций diSхим внутри системы остается без изменений.
Этот вывод следует из приведенных ранее записей. Действительно, если сравнивать, например, закрытые системы без внутренних химических превращений (см. раздел 5.2) и с внутренними химическими превращениями (см. раздел 7.1.), то их принципиальное отличие выражается в записи уравнений изменения внутренней энергии системы (5.24) и (7.1):
без химических превращений
dU = ТdеS – рdеV;
с химическими превращениями
dU = ТdеS – рdеV – ТdiSхим,
или, что то же самое
dU = dQ – рdV; dU = dQ – рdV –dQ/.
Из сопоставления приведенных уравнений вытекает важное следствие, относящееся к изменению функций состояния dU и dН и выражающееся в следующем: для систем без химических превращений знак изменения функций dU и dН совпадает со знаком изменения теплоты системы при ее теплообмене с окружающей средой в соответствии с (5.27) и (5.32)
dU = QV ; dН = QР ,
а для систем с химическими превращениями знак изменения dU и dН противоположен знаку теплового эффекта химической реакции в соответствии с (7.4) и (7.8) без теплообмена системы со средой
![]() ;
;
![]() .
.
Совпадение знаков в этих уравнениях свидетельствует о возможности проведения обратимых процессов при взаимодействии системы с окружающей средой, а несовпадение – о наличии необратимых явлений, связанных с внутренними физико-химическими процессами в системе.
При введении в рассмотрение химической координаты , как независимой переменной, выражения для характеристических функций U, Н, F и G вместо (10.28), (10.38), (10.45) и (10.52) можно записать в виде:
 U
= U(S, V, );
U
= U(S, V, );
Н = Н(S, Р, );
F = F(Т, V, ); (10.110)
G = G(Т, Р, ),
а их полные дифференциалы, как
![]() ;
;
![]() ;
;
(10.111)
![]() ;
;
![]() .
.
Сравнивая эти соотношения с дифференциальными уравнениями (8.33), можно заключить, что последние члены суммы в (10.111) выражают максимальную полезную работу, взятую с обратным знаком.
Частные производные от термодинамических потенциалов по химической переменной, взятые с обратным знаком, при соответствующих (естественных) параметрах, сохраняющих постоянное значение, как раз и определяют термодинамическую величину, называемую химическим сродством:
![]() .
(10.112)
.
(10.112)
Объединенные уравнения первого и второго начал термодинамики по отношению к системам, в которых полезная работа состоит только в изменении их состава, принимают вид
 dU
= ТdS – рdV – А*d
;
dU
= ТdS – рdV – А*d
;
dН = ТdS + Vdр – А*d ;
dF = -SdТ – рdV – А*d ; (10.113)
dG = -SdТ + Vdр – А*d ;
т.е. вместо максимальной полезной работы записывается химическая работа [см. (10.84)].
Из уравнения (10.113) следует, что при постоянстве естественных параметров в системе, в которой протекает химический процесс, изменения всех термодинамических потенциалов равны химической работе и последняя приобретает свойства функции состояния, т.е. становится независимой от пути процесса и определяется только начальным и конечным значениями соответствующего термодинамического потенциала. Например, при постоянных Т и Р химическую работу можно выразить через энергию Гиббса, а при постоянных Т и V – через изменение энергии Гельмгольца системы:
![]()
![]() .
(10.114)
.
(10.114)
Из этого следует также, что химическое сродство А* может выступать в качестве критерия возможности протекания химического процесса в данном направлении и критерия химического равновесия. Сравнение с полученными ранее критериями направленности химических процессов (см. раздел 10.4) дает основание записать следующие соотношения:
d US,V
= -А*d
0
US,V
= -А*d
0
dНS,Р = -А*d 0
dFТ,V = -А*d 0 (10.115)
dGТ,Р = -А*d 0
Так как в направлении протекания химического процесса приращение химической переменной всегда положительно d 0, то критерием возможного протекания процесса при любой паре естественных переменных является положительное химическое сродство, т.е. процесс может идти в данном направлении только в том случае, если химическое сродство положительно, т.е. А* 0.
При наступлении равновесия в системе термодинамические потенциалы принимают постоянное минимальное значение; производная же от них при тех же условиях должна быть равна нулю: А* = 0. Это равенство является общим (при любой паре естественных переменных) условием химического равновесия.
Таким образом, химическое сродство А*, являясь критерием интенсивности протекания химического процесса в данный момент времени, представляет собой также критерий направленности и равновесного состояния:
А*
![]() 0 . (10.116)
0 . (10.116)
Связь между изменением термодинамического потенциала и изменением химического сродства в ходе реакции можно проиллюстрировать, сопоставляя их зависимости от химической переменной .
На рис. 10.2 показано изменение энергии в ходе химической реакции при постоянных давлении и температуре.
Как видно из рисунка, энергия Гиббса уменьшается, а ее производная по химической переменной растет до нуля (она отрицательна). При G=0 в системе устанавливается равновесие и протекание химической реакции прекращается.
Исходя
из соотношений (10.115),
изменение производной
![]() со знаком минус равно химическому
сродству:
со знаком минус равно химическому
сродству:
![]() .
(10.117)
.
(10.117)
С учетом (10.117) связь между G и А* проявляется с помощью рис. 10.3
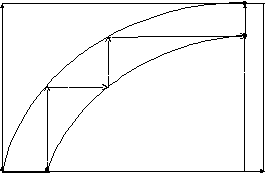
Допустим, что при заданных температуре и давлении, система находится в состоянии, соответствующем точке 1 на рис. 10.3. В системе находятся вещества, способные к самопроизвольному химическому взаимодействию. Эта способность определяется отрицательным значением энергии Гиббса (G0) и положительным значением химического сродства (А*0). Для того, чтобы началось протекание химической реакции, системе необходим «толчок», который может быть создан за счет добавления в нее некоторого количественного избытка одного из исходных веществ. При этом химическая переменная изменяется на величину d, а система выйдет из состояния равновесия (изменение соответствует линии «1-а»). По мере протекания реакции система будет стремиться перейти в новое равновесное состояние, соответствующее точке «в» на рисунке. Этот процесс характеризуется уменьшением отрицательного значения энергии Гиббса (G) и положительного значения химического сродства (А*) – линия «а-в».
G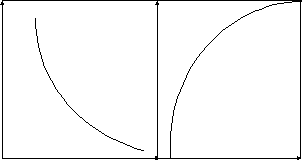
![]() dG=0
dG=0
dG0
Рис. 10.2 Изменение энергии Гиббса системы в ходе химической
реакции при постоянных Т и Р.
Если же продолжить добавление в систему реагента в определенных количествах, то протекание химической реакции продолжится по линии «а-с-е». Этот неравновесный процесс будет продолжаться до тех пор, пока работоспособность системы не будет исчерпана, а система не перейдет в новое равновесное состояние, соответствующее точке 2 (dG=0; А*=0). Вместе с тем этот процесс можно представить как идеализированный равновесный, если выбрать сколь угодно малую скорость и малое количество добавляемого в систему реагента. Другими словами, линия «а-с-е», соответствующая протеканию реального необратимого процесса может быть сколь угодно близка к линии «1-в-d-2», соответствующей идеализированному процессу.
![]() 2
А*=0
2
А*=0
d dG=0 е
dG0
в
с
А*0
1 d а равн
А*=-![]()
Рис. 10.3. Изменения энергии Гиббса в ходе химической реакции при идеализированном обратимом и необратимом процессе (Р,Т=Const).
Из рисунка также видно, что площадь под линией «1-в-d-2» всегда больше площади под кривой «а-с-е». Таким образом, если работа совершается системой, тот работа равновесного процесса имеет большую величину по сравнению с работой неравновесного процесса. Эта потеря работы и принимается в качестве меры необратимого процесса
dАобр – dАнеобр = А*d 0. (10.118)
Теряемая работа оказывается поэтому просто равной некомпенсированной теплоте, и мы снова приходим к основному неравенству де Донде (10.109):
dQ/ = А* d 0.
К такому же выводу можно придти при сопоставлении неравенства (10.118) с соотношением (10.96), вытекающем из уравнений Гиббса-Гельмгольца (10.92-10.94).
Конечные изменения термодинамических потенциалов, взятые с обратным знаком, при постоянных естественных параметрах системы определяют выполненную ей химическую работу при изменении состава системы от 1 до 2 (10.113).
Изменения термодинамических потенциалов, отнесенные к одному пробегу реакции (одному молю реагента или продукта), являются интегральными значениями химического сродства(аналогично интегральным теплотам химического процесса), так же как химическое сродство является дифференциальным значением термодинамического потенциала при постоянстве естественных параметров (10.112).
Покажем расчет интегрального значения химического сродства, исходя из приращения энергии Гиббса при постоянных температуре и давлении (10.113):
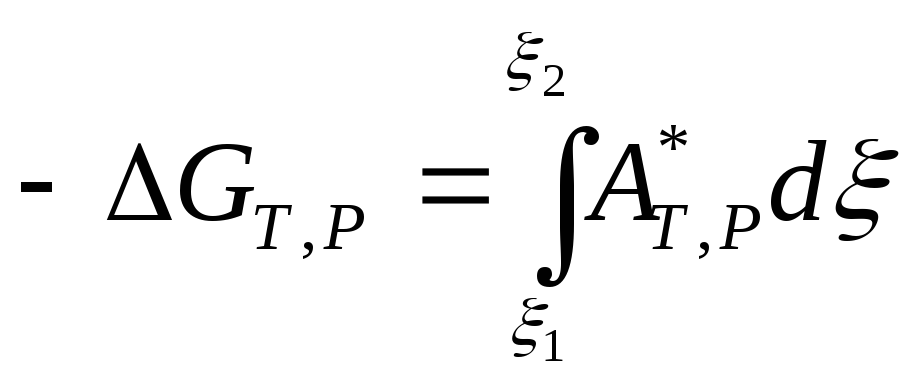 . (10.119)
. (10.119)
Чтобы вычислить интеграл, надо знать зависимость химического сродства А* от химической переменной . Обычно такая зависимость неизвестна. Кроме того, поскольку химическое сродство по мере протекания процесса уменьшается до нуля при достижении равновесия, нельзя говорить даже о приближенном постоянстве химического сродства. Исходя из этого, обычно используют понятие среднего значения химического сродства, соответствующего изменению значений химической переменной от 1 до 2. Это среднее значение при интегрировании принимают постоянным. Тогда решением уравнения (10.119) будет:
![]() .
(10.120)
.
(10.120)
Отсюда находим
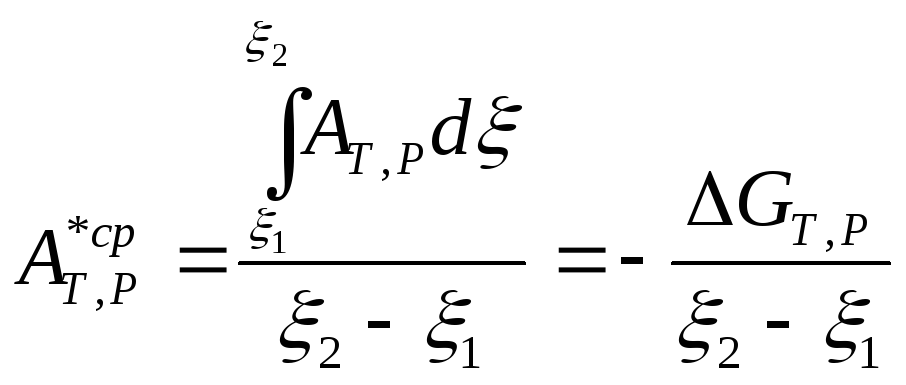 .
(10.121)
.
(10.121)
Если
химический процесс соответствует
протеканию на единицу химической
переменной (т.е. в расчете на один пробег
реакции), то 2
- 1
=1 и среднее значение химического сродства
будет соответствовать удельному
приращению энергии Гиббса (![]() ):
):
![]() (10.122)
(10.122)
(при 2 - 1 =1).
Таким образом, интегральное значение химического сродства, выраженное через изменение энергии Гиббса при постоянных Т и Р и при изменении химической переменной на единицу, отражает среднее значение химического сродства на данном этапе реакции (они численно равны). Из соотношений (10.121) и (10.122) видно, что химическое сродство и изменение энергии Гиббса при постоянных Т и Р можно сопоставить со скоростью движения тела в данный момент и путем, пройденным движущимся телом в единицу времени. Так, среднее химическое сродство в промежутке, равном единице химической переменной (как и средняя скорость движущегося тела), равно изменению энергии Гиббса на протяжении химической переменной (или пути, пройденному телом в единицу времени).
От этой упрощенной аналогии скорости химических превращений со скоростью движущегося тела можно перейти непосредственно к определению кинетики химической реакции. Действительно, контроль над химической переменной ограничен соотношением:
![]() ,
(10.123)
,
(10.123)
где - скорость химической реакции,
t – время.
Согласно неравенству (8.109)
![]() .
(10.124)
.
(10.124)
Это определение кинетики химической реакции также принадлежит де Донде.
При использовании понятия среднего химического сродства неравенство (10.124) можно записать в виде:
![]() ,
(10.125)
,
(10.125)
поскольку
![]() при d=1
в единицу времени равно единице.
при d=1
в единицу времени равно единице.
Из (10.124) или (10.125) вытекает
А *
0 ;
*
0 ;
![]() 0
0
А*
0 ;
![]() 0 (10.126)
0 (10.126)
А* = 0 ; = 0
Соотношение 0 ; А=0 не реализуется, поскольку оно соответствует обратимому протеканию реакции с конечной скоростью, что невозможно.
Таким образом, сродство всегда имеет тот же знак, что и скорость реакции, а при сродстве, равном нулю, скорость реакции также равна нулю, т.е. система находится в состоянии равновесия.
Обратное утверждение, однако, несправедливо. Рассмотрим возможные значения .
Либо
0 , откуда dQ/ 0 и А* 0 ,
что удовлетворяется при
0 ; А* 0
или
0 ; А* 0.
Либо
= 0 , откуда dQ/ = 0 и А* = 0 ,
что удовлетворяется при
= 0 ; А* = 0 (истинное равновесие)
или
= 0 ; А* 0 (ложное равновесие).
Система находится в состоянии ложного равновесия ( или в так называемом заторможенном состоянии), когда реакция в ней не протекает, хотя сродство реакции отлично от нуля. Пример такой реакции графита с кислородом приводился ранее (раздел 10.5). Можно привести и другие примеры, в частности, водород не взаимодействует с кислородом при обычной температуре, хотя сродство реакции велико. Для протекания такой реакции, в соответствии с приведенными рассуждениями, нужно ее инициировать (дать ей «толчок»), что можно показать, вводя в реакционную смесь катализатор или попросту инициируя реакцию с помощью искры.
Исходя из (10.122) можно заключить, что изменение среднего химического сродства равно по величине удельной энергии Гиббса(или удельной энергии Гельмгольца) и противоположно по знаку. Отсюда зависимости химического сродства от температуры, давления и объема прямо противоположны изменению соответствующих термодинамических потенциалов. Химическое сродство должно увеличиваться с ростом температуры, объема и давления, если в процессе соответственно увеличиваются энтропия, давление и уменьшается объем системы при заданной паре постоянных параметров.
Химическое
сродство связано непосредственно с
дифференциальной теплотой реакции
![]() или
или![]() .
Продифференцируем при двух постоянных
параметрах Т и р, Т и V по химической
переменной соотношения (10.49) и (10.42):
.
Продифференцируем при двух постоянных
параметрах Т и р, Т и V по химической
переменной соотношения (10.49) и (10.42):
G = Н – ТS;
F = U – ТS.
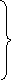 Получим
Получим ![]() ;
;
![]() (10.127)
(10.127)
и ли
ли![]() ;
;
(10.128)
![]()
Из
уравнений (10.127) и (10.128) следует, что
химическое сродство может стать равным
дифференциальной теплоте процесса
![]() или
или![]() при условии малости энтропийного члена,
т.е. при условии
при условии малости энтропийного члена,
т.е. при условии
![]() ,
,
![]() .
.
Такое
условие соблюдается при низких
температурах, или если изменение энтропии
в процессе очень мало, что характерно,
например, для твердофазных реакций. При
этом условии химическое сродство и
теплота реакции имеют, кроме того, разные
знаки, т.е. экзотермические реакции (![]()
0) протекают самопроизвольно (так как
А*
0), что соответствует принципу Бертло.
0) протекают самопроизвольно (так как
А*
0), что соответствует принципу Бертло.
Соотношения (10.128) позволяют получить зависимость химического сродства от температуры с использованием теплоты процесса, что чаще всего применяется в практических расчетах. Такие зависимости, аналогичные уравнениям Гиббса-Гельмгольца, получаются при сопоставлении (10.122) с (10.69) и (10.71). Например:
![]() (10.129)
(10.129)
и
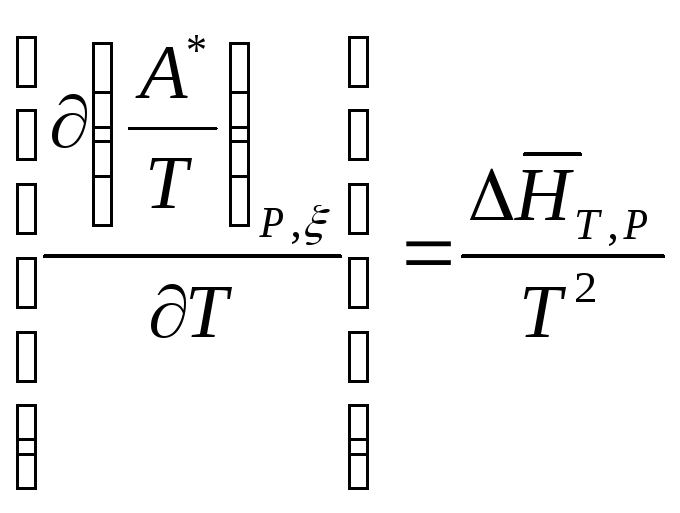 (10.130)
(10.130)
или
![]() (10.131)
(10.131)
и
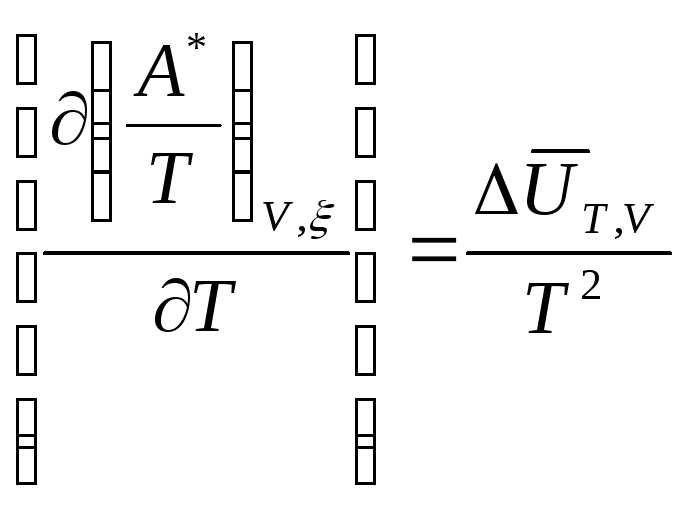 . (10.132)
. (10.132)
Из этих уравнений следует, что химическое сродство увеличивается с повышением температуры, если процесс эндотермический, и уменьшается – если процесс экзотермический.