Интернет-курс Ёршикова С.М. / 5 / 5
.1.doc|
Раздел 5.1 |
Энзимопатии: причины, проявления, методы диагностики. |
|
|
|
|
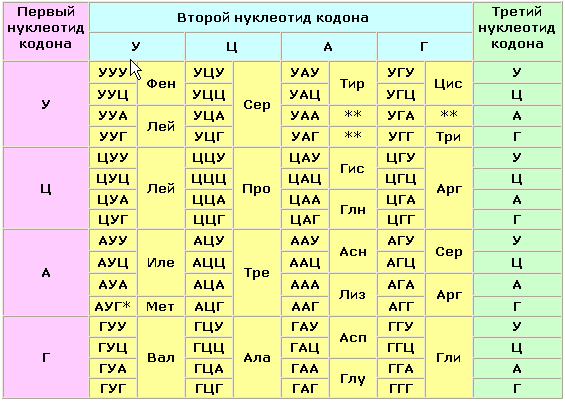
В 1908 году английский врач Арчибальд Гаррод высказал предположение, что причиной ряда заболеваний может являться отсутствие какого-либо из ключевых ферментов, участвующих в обмене веществ. Он ввёл понятие "inborn errors of metabolism" (врождённый дефект обмена веществ). В дальнейшем эта теория была подтверждена новыми данными, полученными в области молекулярной биологии и патологической биохимии. Информация о последовательности аминокислот в полипептидной цепи белка записана в соответствующем участке молекулы ДНК в виде последовательности тринуклеотидных фрагментов - триплетов или кодонов. Каждый триплет кодирует определённую аминокислоту. Такое соответствие называется генетическим кодом. Причём некоторые аминокислоты могут быть закодированы при помощи нескольких кодонов. Существуют также специальные кодоны, являющиеся сигналами для начала синтеза полипептидной цепи и его прекращения. К настоящему времени генетический код полностью расшифрован (таблица 5.1). Он является универсальным для всех видов живых организмов. Таблица 5.1 Триплетный код нуклеотидов мРНК для аминокислот
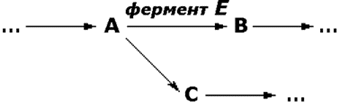
Примечания. * - этот кодон является также сигналом для начала синтеза полипептидной цепи (инициирующий кодон); ** - эти кодоны не соответствуют ни одной из аминокислот и служат сигналом для прекращения синтеза полипептидной цепи (терминирующие кодоны). Реализация информации, заложенной в молекуле ДНК, включает несколько этапов. Сначала в клеточном ядре в процессе транскрипции синтезируется матричная РНК (мРНК), поступающая в цитоплазму. В свою очередь, мРНК служит матрицей для трансляции - синтеза полипептидных цепей на рибосомах. Таким образом, природа молекулярных болезней определяется нарушением структуры и функции нуклеиновых кислот и контролируемых ими белков. МутацииПоскольку информация о структуре всех белков клетки содержится в последовательности нуклеотидов ДНК, а каждая аминокислота определяется триплетом нуклеотидов, изменение первичной структуры ДНК может в конечном счёте оказать глубокое влияние на синтезируемый белок. Подобные изменения происходят за счёт ошибок репликации ДНК, когда одно азотистое основание заменяется другим, либо в результате действия радиации или при химической модификации. Все возникшие таким образом наследуемые дефекты называются мутациями. Они могут приводить к неправильному считыванию кода и делеции (выпадению) ключевой аминокислоты, замене одной аминокислоты другой, преждевременной остановке белкового синтеза или добавлению аминокислотных последовательностей. Учитывая зависимость пространственной упаковки белка от линейной последовательности в нём аминокислот, можно полагать, что подобные дефекты способны изменить структуру белка, а значит, и его функцию. Тем не менее, многие мутации обнаруживаются только в лабораторных условиях и не оказывают вредного воздействия на функции белка. Таким образом, ключевым моментом является локализация изменений в первичной структуре. Если положение замененной аминокислоты окажется критическим для формирования третичной структуры и образования каталитического центра фермента, то мутация является серьёзной и может проявиться как заболевание. Последствия для обмена веществПоследствия недостаточности одного фермента в цепи реакций обмена веществ могут проявляться по-разному. Предположим, что превращение соединения A в соединение B катализирует фермент Е и что соединение C встречается на альтернативном пути превращений (рисунок 5.1):
Рисунок 5.1. Схема альтернативных путей биохимических превращений. Последствиями недостаточности фермента могут быть следующие явления:
Если метаболическое превращение в целом регулируется по принципу обратной связи конечным продуктом, то эффекты двух последних типов аномалий будут более значительными. Так, например, при порфириях (врождённых нарушениях синтеза гема) устраняется подавляющего эффекта гема на начальные реакции синтеза, что приводит к образованию избыточных количеств промежуточных продуктов метаболического пути, которые обладают токсическим действием на клетки кожи и нервной системы. Факторы внешней среды могут усиливать или даже полностью определять клинические проявления некоторых врождённых нарушений обмена веществ. Например, у многих пациентов с недостаточностью глюкозо-6-фосфатдегидрогеназы заболевание начинается только после приёма таких лекарственных средств, как примахин. В отсутствие контактов с лекарственными средствами такие люди производят впечатление здоровых. Лабораторная диагностика врождённых нарушений обмена веществО недостаточности фермента обычно судят косвенно по повышению концентрации исходного вещества, которое в норме подвергается превращениям под действием данного фермента (например, фенилаланин при фенилкетонурии). Прямое определение активности таких ферментов проводят только в специализированных центрах, но по возможности диагноз следует подтверждать этим методом. Пренатальная (дородовая) диагностика некоторых врождённых нарушений метаболизма возможна путём иследования клеток амниотической жидкости, полученных на ранних стадиях беременности и культивируемых in vitro. Лечение при врождённых нарушениях метаболизмаНекоторые врождённые нарушения метаболизма поддаются лечению путём доставки в организм недостающего метаболита или путём ограничения поступления в желудочно-кишечный тракт предшественников нарушенных процессов обмена веществ. Иногда могут быть удалены накапливающиеся продукты (например, железо при гемохроматозе). |
|
|
© С.М.Ершиков, 2007. Все права защищены |
|