

Физика 2009 (F) / МОЛ-ТЕРМ / Упр2_МД
.doc«ТЕПЛОВОЕ ДВИЖЕНИЕ МОЛЕКУЛ»
КГТУ. Каф. Физики. Гайсин Н.К., Казанцев С.А., Минкин В.С., Самигуллин Ф.М.
Для перемещения по тексту можно использовать:
1- нажатие клавиш PgDn, PgUp,, для перемещения по страницам и по строкам;
2- нажатие левой клавиши «мыши» по выделенному тексту для перехода в требуемый раздел;
3- нажатие левой клавиши «мыши» по выделенному значку @ для перехода в оглавление
ОГЛАВЛЕНИЕ
-
Характер теплового движения молекул в разных состояниях. Средние энергии молекул в разных фазах. Распределение молекул по скоростям.
-
Диффузия. Коэффициент диффузии.
-
Моделирование движения молекул с помощью компьютера.
-
Упражнение . Наблюдение и анализ: 1-траекторий движения молекул в трех агрегатных состояниях, 2- графиков распределения молекул по скоростям, 3-радиальных функций распределения, 4-коэффициентов диффузии.
@ 1. .Характер теплового движения молекул в разных состояниях. Средние энергии молекул в разных фазах. Распределение молекул по скоростям.
Как известно молекулы и атомы в веществе постоянно находятся в движении, которое имеет случайный, хаотический характер. Тем не менее в каждом агрегатном состоянии имеются характерные особенности этого движения, которые во многом определяют свойства различных состояний. Это связано с тем, что межмолекулярные силы взаимодействия стремятся сблизить молекулы, а тепловое хаотичное движение препятствует этому и такие две тенденции в разных агрегатных состояниях дают, значительно отличающиеся, вклады в характер движения молекул. Для количественного анализа влияния различных вкладов, обычно рассматривают величину полной средней энергии молекулы и вклад в эту энергию кинетической и потенциальной составляющих.
В газах среднее расстояние между молекулами больше их размеров, силы притяжения малы, а интенсивность движения значительна, что не позволяет молекулам объединиться на длительное время, а при отсутствии сосуда молекулы стремятся заполнить все доступное пространство. В газах потенциальная энергия взаимодействия отрицательна, кинетическая энергия имеет большую величину, поэтому полная энергия молекулы положительна и при расширении молекулярная система может совершать работу над внешними системами. Вследствие этого, молекулы распределены в пространстве равномерно, большее время находятся на больших расстояниях (Рис.1а) и двигаются равномерно и прямолинейно без взаимодействия. Взаимодейтвие молекул имеет кратковременный характер и происходит только при их столкновении, что приводит к значительному изменению траектории движения.
В твердых телах среднее расстояние между молекулами сравнимо с их размерами, поэтому силы притяжения очень велики и даже сравнительно большая интенсивность движения не позволяет молекулам разойтись на большие расстояния. В данном случае отрицательная потенциальная энергия взаимодействия много больше кинетической энергии, поэтому полная энергия молекулы также отрицательна и для разрушения твердого тела необходимо совершать значительную работу. Молекулы в твердом теле располагаются на строго определенных расстояниях друг от друга и совершают колебательные движения около некоторых средних положений, называемых узлами кристаллической решетки (Рис.1в).
В жидкостях расстояние между молекулами сравнимо с их размерами, силы притяжения велики, но интенсивность теплового движения тоже большая, что позволяет молекулам по истечении некоторого времени отойти друг от друга на большие расстояния. В жидкостях отрицательная потенциальная энергия взаимодействия сравнима по величине с кинетической энергией, поэтому полная энергия молекулы близка к нулю, что позволяет жидкости легко деформироваться и без разъединения занимать доступный объем под действием даже слабых внешних сил. Молекулы в жидкости, находятся в среднем на определенных, близких друг от друга расстояниях и совершают, похожие на колебания, движения около средних положений, которые также перемещаются хаотически в пространстве (Рис.1б).

Рис. 1. Характер движения молекул в газах (а), жидкостях (б) и твердых (в) телах
В результате взаимодействия между молекулами молекулярная система через некоторое время, называемым временем релаксации, приходит в равновесное состояние, характеризуемое: 1- определенным уравнением состояния, связывающим термодинамические параметры вещества; 2- определенной радиальной функцией, характеризующей распределения молекул в пространстве; 3- функцией Максвелла, характеризующей распределения молекул по скоростям (Рис.2).
При каждом акте взаимодействия молекул друг с другом их скорости меняются и в результате через некоторое время устанавливается равновесное состояние, при котором число молекул dN, имеющих скорость в определенном диапазоне значений dVсохраняется постоянным и определяется функцией Максвелла F(V) согласно соотношениям
dN = N F(V)V, F(V)=4V2(m/2kT)3/2 exp(-mV2/2kT).
Вид этой функции показан на Рис.2, она существенно зависит от температуры Т и характеризуется наличием максимума, который указывает на наличие наиболее вероятной скорости Vвер. Как видно из графиков (Рис.2), в веществе имеются молекулы с любыми скоростями, но число молекул со скоростями в диапозоне dV около наивероятнейшей будет наибольшим. Максвелловское распределение молекул по скоростям характерно для всех агрегатных состояний, но время релаксации к такому распределению у них разное, это связано с различием времени взаимодействия молекул в разных фазах.
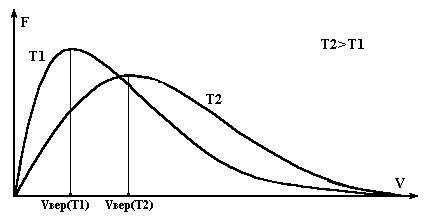 Рис.
2.
Максвелловское распределение молекул
по скоростям.
Рис.
2.
Максвелловское распределение молекул
по скоростям.
@2_Диффузия. Коэффициент диффузии.
Вследствие теплового движения молекул в веществе происходит диффузия. Диффузия это явление переноса вещества из одной части занимаемого им объема в другую. Это явление наиболее сильно проявляется в газах и жидкостях, в которых тепловое движение молекул особенно интенсивно и возможно на большие расстояния.
Феноменологически диффузия описывается законом Фика, который устанавливает связь между удельным потоком Ji компонента i и градиентом концентрации этого компонента вещества ni
![]() .
.
Удельный поток диффузии Ji - это количество молекул компонента i, перенесенного за единицу времени через единицу площади поперечного сечения, перпендикулярного к направлению потока вещества, ni – числовая плотность компонента i, Di – коэффициент диффузии, V0 – гидродинамическая скорость вещества. Коэффициент диффузии в системе СИ имеет размерность м2с-1. Знак минус в формуле Фика указывает на то, что поток диффузии направлен обратно направлению роста концентрации вещества. Уравнение Фика описывает только стационарный процесс диффузии, при котором концентрация, ее градиент и диффузионный поток не зависят от времени.
![]() Механизм
диффузии в газах подробно рассматривается
в разделе молекулярной физики.
Молекулярно-кинетическая теория газов
приводит к известному выражению для
коэффициента диффузии
Механизм
диффузии в газах подробно рассматривается
в разделе молекулярной физики.
Молекулярно-кинетическая теория газов
приводит к известному выражению для
коэффициента диффузии
 ,
,
где
![]() i-
средняя длина свободного пробега и
i-
средняя длина свободного пробега и
![]() i-
средняя арифметическая скорость
поступательного движения молекул газа
сорта i,
di
– эффективный диаметр, mi
- масса молекул, ni
– числовая плотность, p
– давление. Эта формула соблюдается в
довольно широком интервале давлений и
температур для не плотных газов и дает
значение порядка 10-5
м2/c.
i-
средняя арифметическая скорость
поступательного движения молекул газа
сорта i,
di
– эффективный диаметр, mi
- масса молекул, ni
– числовая плотность, p
– давление. Эта формула соблюдается в
довольно широком интервале давлений и
температур для не плотных газов и дает
значение порядка 10-5
м2/c.
Однако диффузия молекул в жидкостях значительно отличается от диффузии в газах, это связано с различием характера движения молекул в этих фазах. Плотность вещества в жидком состоянии в тысячи раз больше его плотности в газообразном состоянии. Поэтому в жидкостях каждая молекула сидит в плотном окружении соседних молекул и не имеет свободы поступательных перемещений как в газах. Согласно известной теории Френкеля, молекулы в жидкостях, как и в твердых телах, совершают беспорядочные колебания около положений равновесия. Эти положения можно рассматривать как потенциальные ямы, созданные окружающими молекулами. В кристаллах молекулы не могут покидать свои положения равновесия, и поэтому можно считать, что в них практически отсутствуют поступательные перемещения молекул. В жидкостях такие положения не являются постоянными. Время от времени молекулы меняют свои положения равновесия, оставаясь в плотном окружении других молекул.
Диффузию
молекул в однокомпонентных жидкостях,
обусловленную их тепловым движением в
отсутствие градиентов концентрации,
обычно называют самодиффузией молекул.
Для того, чтобы молекулы, преодолев
взаимодействие с окружающими молекулами,
могли совершить переход в новое положение,
необходима энергия. Та минимальная
энергия, которая необходима, чтобы
молекула покинула временную потенциальную
яму, называется энергией активации.
Молекулу, получившую такую энергию,
называют активированной. Молекулы,
совершающие беспорядочные колебания,
активируются в результате столкновений
с окружающими молекулами. Энергия
активации в жидкостях значительно
меньше, чем в кристаллах. Поэтому переходы
молекул в жидкостях с одного места на
другое значительно чаще, чем в кристаллах.
Число активированных молекул определяется
распределением Больцмана, и частота
переходов (скачков㿹
молекул
в новые положения ,
определяется приближенной формулой
![]() ,
где 0
-
коэффициент, слабо зависящий от
температуры, Е
– энергия активации.
,
где 0
-
коэффициент, слабо зависящий от
температуры, Е
– энергия активации.
Для
получения формулы коэффициента диффузии
для жидкости рассмотрим диффузионный
поток через некоторую поверхность
площадью s.
При тепловом движении молекулы проходят
через эту поверхность как в прямом, так
и в обратном направлениях. Поэтому
удельный диффузионный поток может быть
выражен в виде
![]() ,
где знаки соответствуют прямому и
обратному направлению оси х.
Найдем величины J+
и J--
. Через
выделенную поверхность, очевидно, за
один скачок могут пройти без отклонения
только те молекулы, которые находятся
от нее на расстоянии не далее средней
длины скачка молекул δ.
Построим по обе стороны от поверхности
цилиндр с площадью основания s.
Через поверхность s
пройдут только те молекулы, которые
заключены в объеме цилиндра δs.
Однако пройдут не все молекулы, а только
те из них скачки которых направлены
вдоль оси х.
Если считать, что молекулы с равной
вероятностью движутся вдоль осей х, у
и z,
то через сечение в данном направлении
пройдет только 1/6 часть общего числа
молекул в цилиндре. Тогда число молекул,
проходящих за один скачок через
поверхность s
в прямом направлении N+
выразится
в виде
,
где знаки соответствуют прямому и
обратному направлению оси х.
Найдем величины J+
и J--
. Через
выделенную поверхность, очевидно, за
один скачок могут пройти без отклонения
только те молекулы, которые находятся
от нее на расстоянии не далее средней
длины скачка молекул δ.
Построим по обе стороны от поверхности
цилиндр с площадью основания s.
Через поверхность s
пройдут только те молекулы, которые
заключены в объеме цилиндра δs.
Однако пройдут не все молекулы, а только
те из них скачки которых направлены
вдоль оси х.
Если считать, что молекулы с равной
вероятностью движутся вдоль осей х, у
и z,
то через сечение в данном направлении
пройдет только 1/6 часть общего числа
молекул в цилиндре. Тогда число молекул,
проходящих за один скачок через
поверхность s
в прямом направлении N+
выразится
в виде
![]() ,
где
n1
– число
молекул в единице объема на расстоянии
δ
влево от поверхности s.
Аналогичное рассуждение о прохождении
молекул через поверхность s
в обратном направлении приведет к
выражению
,
где
n1
– число
молекул в единице объема на расстоянии
δ
влево от поверхности s.
Аналогичное рассуждение о прохождении
молекул через поверхность s
в обратном направлении приведет к
выражению
![]() ,
где n2
– число
молекул в единице объема на расстоянии
δ
вправо от поверхности s.
Тогда диффузионные потоки могут быть
найдены как
,
где n2
– число
молекул в единице объема на расстоянии
δ
вправо от поверхности s.
Тогда диффузионные потоки могут быть
найдены как
![]() и
и
![]() .
Полный поток выразится в виде
.
Полный поток выразится в виде
![]()
![]() ,
где n1-n2
– разность концентраций молекул в
слоях, отстоящих друг от друга на среднем
расстоянии
δ можно
записать в виде n1-n2=nx.
Тогда получаем
,
где n1-n2
– разность концентраций молекул в
слоях, отстоящих друг от друга на среднем
расстоянии
δ можно
записать в виде n1-n2=nx.
Тогда получаем
![]() .
Сравнивая эту формулу с законом Фика
для случая когда V0=0,
находим
.
Сравнивая эту формулу с законом Фика
для случая когда V0=0,
находим
![]() ,
,
откуда
![]() ,
где
,
где
![]() - коэффициент, слабо зависящий от
температуры, эта формула для жидкостей
и плотных газов дает значение для D
порядка 10-9
м2/c.
- коэффициент, слабо зависящий от
температуры, эта формула для жидкостей
и плотных газов дает значение для D
порядка 10-9
м2/c.
Явление
самодиффузии молекул можно также
анализировать путем рассмотрения
теплового поступательного движения
молекул, как серии беспорядочных,
равновероятных перемещений (блужданий).
За некоторый достаточно большой
промежуток времени молекулы могут
описать длинную траекторию, однако они
сместятся от первоначального положения
на незначительное расстояние. Рассмотрим
совокупность молекул в виде беспорядочно
движущихся частиц, выберем из этой
совокупности некоторую молекулу и
предположим, что она в начальный момент
времени находится в начале системы
координат. Далее через равные промежутки
времени Δt
будем отмечать радиусы-векторы ее места
нахождения r(ti).
Вектор перемещения молекулы между
(i-1)-м
i
–м моментами времени будет выражаться
в виде Δri
= r(ti)-r(ti-1).
К
моменту времени tк
= k
Δt
молекула окажется смещенной из начальной
точки наблюдения в точку с радиусом-вектором
r(tк),
который выражается как векторная сумма
смещений
![]() r(tк)
=
r(tк)
=
![]() ri.
Квадрат смещения частицы за это время
выразится в виде
ri.
Квадрат смещения частицы за это время
выразится в виде
r(tк)
= (![]() Δri)2
=
Δri)2
=![]()
![]() (ΔriΔrj)
+
(ΔriΔrj)
+
![]() Δri2
.
Δri2
.
Усредним
полученное выражение по всем молекулам
рассматриваемой совокупности, тогда
ввиду независимости смещений молекул
в разные промежутки времени в двойной
сумме одинаково часто встречаются как
положительные так и отрицательные
значения скалярного произведения,
поэтому ее статистическое среднее равно
нулю. Тогда средний квадрат смещения
частиц запишется как <r2(tk)>
=
![]() <Δri2>.
В жидкости <Δri2>
следует считать равным среднему квадрату
скачка молекул δ2, а число
скачков за время tk
равным tk
. Тогда <r2(tk)>=tk
δ2.
Сопоставив это выражение с формулой
для D, получаем известное
соотношение Эйнштейна, из которого
становится ясным молекулярно-кинетический
смысл коэффициента диффузии D
<Δri2>.
В жидкости <Δri2>
следует считать равным среднему квадрату
скачка молекул δ2, а число
скачков за время tk
равным tk
. Тогда <r2(tk)>=tk
δ2.
Сопоставив это выражение с формулой
для D, получаем известное
соотношение Эйнштейна, из которого
становится ясным молекулярно-кинетический
смысл коэффициента диффузии D
<r2(t)> = 6Dt.
Можно доказать, что коэффициенты диффузии в формулах Эйнштейна и Фика идентичны. Для однокомпонентной системы этот коэффициент называют коэффициентом самодиффузии, в случае диффузии в многокомпонентных смесях при наличии у них градиентов концентраций, потоки отдельных компонентов можно определить если известны коэффициенты диффузии всех компонентов в смеси. Экспериментально их находят методам радиоактивных меток или методом ядерного магнитного резонанса, в которых можно определять средний квадрат смещения «меченных» молекул.
@3_Моделирование движения молекул с помощью компьютера.
Современные средства вычислительной техники обладают огромной памятью и высоким быстродействием. Такие качества делают их незаменимым средством моделирования ряда физических процессов. В молекулярной физике широко развит метод молекулярной динамики - метод моделирования молекулярного движения. Этот метод широко применяется в газах, жидкостях, кристаллах и полимерах. Он сводится к численному решению уравнений динамики движения частиц в ограниченном объеме пространства с учетом взаимодействий между ними и может имитировать поведение молекул в произвольных условиях, аналогичным реальным. В этом отношении можно его уподобить реальному эксперименту, поэтому такое моделирование иногда называют численным экспериментом. Значение этих “экспериментов” состоит в том, что они дают возможность следить во времени за изменением нескольких макроскопических параметров, характеризующих систему частиц, и усредняя их по времени или по числу частиц, получать термодинамические параметры моделируемых реальных систем. Кроме того, они дают возможность визуализации молекулярного движения, позволяя следить за траекторией любой отдельно взятой частицы.
Алгоритм моделирования состоит из нескольких этапов. Вначале определенное число частиц (в пределах 102-103) хаотично распределяют в некотором ограниченном объеме (в ячейке), задавая случайным образом начальные скорости и координаты каждой частицы. Начальные скорости частиц задаются так, чтобы средняя кинетическая энергия поступательного движения частиц была равна (3/2)кТ, т.е. соответствовала температуре опыта, а начальные координаты задают в соответствии со средним межмолекулярным расстоянием моделируемой системы.
Далее, зная потенциал взаимодействия частиц (например, потенциал Леннарда-Джонса) и соответственно силу межмолекулярного взаимодействия производится расчет результирующих мгновенных сил, действующих на каждую частицу со стороны всех остальных частиц, и по уравнению динамики (второму закону Ньютона) вычисляются мгновенные ускорения частиц, вызванные, действием этих сил. Зная ускорения, а также начальные координаты и скорости, производится расчет скоростей и координат частиц в конце заданного малого промежутка времени t (обычно 10-14 с). При средней скорости движения частиц около 103 м/с, смещение частиц за такой малый промежуток времени составляет величину порядка 10-11м, что значительно меньше их размеров.
Последовательное повторение таких вычислений с запоминанием мгновенных сил, скоростей и координат частиц, позволяет знать координаты и скорости всей системы частиц на достаточно большом промежутке времени. Ограниченность объема учитывается специальными граничными условиями. Либо полагают, что на границе заданного объема частица испытывает абсолютно-упругое соударение со стенкой и вновь возвращается в объем, либо считают, что данная ячейка окружена со всех сторон такими же ячейками и, если частица выходит из данной ячейки, то одновременно тождественная ей частица входит с противоположной ячейки. Таким образом, число частиц и их полная энергия в объеме ячейки не меняются. Вследствие математически случайного характера первоначального распределения частиц по скоростям и координатам, необходимо некоторое время (время релаксации –10-12- 10-11 с), в течении которого в системе устанавливается равновесное состояние частиц по скоростям (Максвелловское распределение скоростей) и по координатам (распределение в соответствии с радиальной функцией распределения).
Значения макроскопических параметров, характеризующих систему, вычисляются путем их усреднения по траектории или по скоростям частиц. Например, давление на стенки сосуда можно получить путем усреднения изменений импульсов частиц, сталкивающихся с границами ячейки. Усреднением числа частиц в шаровых слоях, находящихся на различных расстояниях r от выбранной молекулы, можно определить радиальную функцию распределения. По средним квадратам смещений частиц за заданное время, можно рассчитать коэффициенты самодиффузии молекул. Подобным образом определяют и другие искомые характеристики.
Естественно, процессы, происходящие в системе частиц за короткое время, рассчитываются ЭВМ за значительное время. Машинное время, затрачиваемое на расчеты, может составить десятки, а то и сотни часов. Это зависит от числа выбранных в ячейке частиц и от быстродействия ЭВМ. Современные ЭВМ позволяют моделировать динамику до 104 частиц, доводя время наблюдения за процессом их перемещений до 10-9с , точность расчета характеристик исследуемых систем позволяет не только уточнять теоретические положения, но и использовать их на практике.
@4_Упражнение. Наблюдение и анализ: 1-траекторий движения молекул в трех агрегатных состояниях, 2- графиков распределения молекул по скоростям, 3-радиальных функций распределения, 4-коэффициентов самодиффузии.
В данном упражнении программа компьютера моделирует методом молекулярной динамики движение атомов аргона (с потенциалом взаимодействия Леннард-Джонса) в трех агрегатных состояниях: плотный газ, жидкость, твердое тело. Для выполнения данного упражнения необходимо войти в программу MD-L4.EXE, последовательно просмотреть и выполнить предлагаемые пункты меню.
Меню программы содержит четыре пункта:
1 ИНСТРУКЦИЯ ДЛЯ РAБОТ Ы,
2 ВЫБОР ПAРAМЕТРОВ МОДЕЛИРУЕМЫХ СОСТОЯНИЙ,
3 МОДЕЛИРОВAНИЕ ДИНAМИКИ ЧAСТИЦ,
4 КОНЕЦ РAБОТЫ.
В пункте 1- <<ИНСТРУКЦИЯ ДЛЯ РAБОТЫ>> рассказывается о программе и о методике работы с программой. Необходимо отметить и запомнить: 1) Данная программа предусматривает работу в двух режимах для выполнения двух видов работ, необходимых при моделировании молекулярного движения в разных фазах; 2) Результаты моделирования выдаются на два экрана, переключение между которыми производится одновременным нажатием клавиш Alt+1 и Alt+2, останов работы программы и выход в меню производится одновременным нажатием клавиш Ctrl и S; 3) Для правильного выполнения программы необходимо следить за ее сообщениями и правильно выполнять их.
В пункте 2 программа работает в режиме <<ВЫБОР ПAРAМЕТРОВ МОДЕЛИРУЕМЫХ СОСТОЯНИЙ>> , который позволяет рассмотреть фазовую диаграмму для системы частиц с потенциалом взаимодействия Леннард-Джонса [D.M.Heyes,J.Chem.Phys., N96(V3), p2217, 1992] и рассчитать для различных агрегатных состояний следующие параметры: приведенное давление P*=Pd3/e и приведенную полную энергию одной частицы U*=u/e. 3десь: n-числовая плотность, u-внутренняя энергия одной частицы, к-постоянная Больцмана, P-давление, Т-температура, d-эффективный диаметр частицы, е-глубина потенциальной ямы. Для расчета необходимо рассмотреть фазовую диаграмму в координатах n*, T* (n*=nd3 - приведенная числовая плотность, T*=кT/e - приведенная температура) и ввести n*, T*. На этой фазовой диаграмме Вам необходимо найти области: плотного газа, жидкого, твердого состояний и ввести n*, T* для трех точек в каждой из этих областей. Для анализа влияния температуры необходимо выбирать точки с разными температурами, но с одинаковыми плотностями (T* и n* можно взять из Таблицы N1). Выбранные Вами и рассчитанные программой, термодинамические параметры этих точек для трех состояний занесите в Таблицу N1, для этих точек Вы будете проводить моделирование движения атомов аргона.
В пункте меню 3 программа работает в режиме <<МОДЕЛИРОВAНИЕ ДИНAМИКИ ЧAСТИЦ>> , он позволяет рассматривать картину движения молекул в разных агрегатных состояниях и рассчитывать путем усреднения ряд термодинамических параметров. После выбора (с помощью дополнительного меню) типа моделируемого агрегатного состояния (плотный газ, жидкость, твердое тело), программа предложит Вам параметры этого состояния, заложенные в программу, если Вы выбрали другие параметры, то их можно на данном этапе изменить согласно Таблице N1 (для этого на запрос <<ВЫ БУДЕТЕ МЕНЯТЬ ПЛОТНОСТЬ И ТЕМПЕРAТУРУ ? (Y/N)>> нажмите Y, в противном нажмите N) . В данном режиме информация о динамике выдается на два экрана, для включения которых необходимо нажать Alt и 1 или Alt и 2.
На первый экран выводятся данные о системе и графики флюктуаций для: 1-температуры, 2-потенциальной энергии частицы, 3-кинетической энергии, 4-полной энергии частицы. Кроме этого в бегущей строке выдается мгновенная дополнительная численная информация: Ni-текущее число шагов итераций, t(c)-физическое время моделирования динамики, ЕР+ЕК(Дж)-полная энергия одной частицы, U*-приведенная энергия, Т(К)-температура, ti(c)-машинное время счета одного шага для одной частицы, P*-приведенное давление, Pv(Па)-давление(вериал), P=nkT, dt(c)-шаг интегрирования по времени.
На второй экран выводятся траектории частиц и графики характеристик, полученные путем усреднения динамических параметров движения частиц: 1-графики распределения частиц по скоростям на фоне распределения Максвелла (Vвер - наиболе вероятная скорость, температуры заданные и средняя); 2-график радиальной функци распределения, 3-график зависимости среднего квадрата смещения частиц от времени и значение коэффициента самодиффузии.
После запуска программы Вам необходимо наблюдать за изменениями характеристик и дождаться момента времени, когда флюктуации потенциальной и кинетической станут достаточно малы (5-10%). Это состояние можно считать равновесным, оно достигается программой автоматически путем проведения динамики в течении 2.10-12c, после этого радиальная функция распределения и функция распределения по скоростям будут соответствовать равновесным. После достижения равновесного состояния (примерно через 1.10-11 с.) необходимо занести требуемые данные с обоих экранов в Таблицу N2. Аналогичные расчеты провести для трех температур в каждом агрегатном состоянии, для последней температуры зарисовать функцию распределения по скоростям и радиальную функцию распределения.
После окончания работы через пункт 4- <<КОНЕЦ РAБОТЫ>> необходимо вернуться к работе с методическим пособием.
Приготовьте в своей тетрадке Таблицу N1, Таблицу N2 .