

кванты лекции
.pdfинтерпретировать свойства известных систем, но и моделировать новые соединения с наперед заданными свойствами и предсказывать их реакционную способность.
Схема сравнения результатов квантово-химических расчетов с данными измерений по методам термохимии и кинетики для реакций между простыми молекулами состоит в следующем. Прежде всего, принимают приближение Борна – Оппенгеймера, в результате чего уравнение Шрёдингера для электронно-ядерной системы распадается на уравнение Шрёдингера для электронов и уравнение Шрёдингера для ядер. Колебательную задачу, состоящую в решении уравнения Шрёдингера для ядер, опускают из рассмотрения. Совокупность решений уравнения Шрёдингера для электронов при различных конфигурациях ядерной подсистемы определяет ППЭ химической реакции как совокупность точек, характеризуемых определенной энергией системы при фиксированном положении ядер. В результате получаются потенциальная энергия основного состояния реагирующей системы и электронная волновая функция.
Расчет особых точек ППЭ для элементарного акта осуществляется в предположении, что реагентам и продуктам соответствуют локальные минимумы, а переходному состоянию – седловая точка ППЭ. В этих точках выполняются условия стационарности (5). В минимуме все собственные значения матрицы Гессе (6) положительны. Диагонализированная матрица Гессе (7) имеет в седловой точке переходного состояния (одномерном локальном максимуме) на ППЭ одно- единственное отрицательное значение.
Определенные трудности вызывает определение пути химической реакции. Если локализация переходного состояния неоднозначна (т.е. неизвестна точная геометрия активированного комплекса), то, очевидно, можно построить много путей, соединяющих соседние минимумы ППЭ через седловую точку. Согласно классическому определению Эйринга и Поляни путь химической реакции есть путь минимальной энергии. Его также можно определить как путь наискорейшего спуска и т. д. В настоящее время путь химической реакции обычно определяют как путь, задаваемый внутренней координатой реакции в базисе масс-взвешенных координат, которые определяются как
Qi = ∑ai mi1 / 2αi , |
(104) |
i |
|
где αi = x, y, z; ai – коэффициенты разложения внутренней координаты реакции по декартовым. Внутренняя координата реакции Q есть путь минимальной энергии, проходящий через долину реагентов, долину продуктов и точку переходного состояния.
Полуколичественная оценка геометрии переходного состояния химической реакции в случае высокой симметрии реагирующей системы может быть осуществлена с использованием доказательства единственности координаты реакции. Если форма ППЭ близка к параболической, для определения геометрии переходного состояния можно воспользоваться постулатом Хэммонда, связывающим геометрию переходного состояния с тепловым эффектом реакции. По Хэммонду экзотермическим реакциям соответствует реагентоподобное переходное состояние, эндотермическим – продуктоподобное, теплонейтральным – лежащее примерно по середине между реагентами и продуктами. Кроме того, используются модельные подходы, имеющие в качестве квантово-химической основы орбитальные представления (правила отбора) и теорию возмущений (метод индексов реакционной способности, теория граничных орбиталей).
5.3. Правила отбора для осуществимости реакций
В различных превращениях электронные состояния химических систем стремятся оставаться неизменными, в частности, сохранить имеющиеся у нее признаки симметрии: суммарный спин состояния (S), орбитальный момент (Λ), а также орбитальную симметрию рвущихся и образующихся связей. Несохранение этих признаков означает «запрет» по симметрии на протекание реакции. Однако запрет по симметрии не означает невозможности протекания реакции. Как правило, запрещенные по симметрии реакции требуют для их осуществления высокой энергии активации.
4
5.3.1. Запрет по спину (первое правило Вигнера – Витмера)
Реакция запрещена по спину, если среди спиновых состояний продуктов нет ни одного состояния, совпадающего с одним из спиновых состояний исходных веществ. Например, реакция
образования молекулы кислорода из атомов разрешена по спину: O (3P) + O (3P) → O2 (3Σ).
Обозначим спиновые моменты атомов кислорода S1 и S2. По правилу сложения моментов суммарный спин двух атомов кислорода может принимать следующие значения (S1 ≥ S2): S1 + S2, S1 + S2 – 1, …, S1 – S2 + 1, S1 – S2. Так как для атомов кислорода S1 = S2 = 1, то возможные значения суммарных спинов 2, 1 и 0. Спин продукта реакции (S3 = 1) совпадает с одним из возможных значений спина исходных веществ. Следовательно, реакция разрешена по спину. Эта реакция не имеет других запретов и, как показывает опыт, протекает без потенциального барьера.
Аналогичный вывод можно сделать для реакции
O (3P) + H (2S) → OH (2Π).
Так как S1 = 1, S2 = 1/2, то их суммарный спин может принимать значения 3/2 и 1/2, одно из которых совпадает с S3 = 1/2. Из экспериментальных данных следует, что эта реакция также протекает при энергии активации, равной нулю.
В противоположность этому реакция
N2O (1Σ) → N2 (1Σ) + O (3P)
запрещена по спину. Действительно, спин исходного вещества S1 = 0, сумма же спинов продуктов реакции S2 = 0 и S3 = 1 принимает только одно значение, равное 1. Так как спины исходных веществ и продуктов не совпадают, то реакция «запрещена». Фактически эта реакция протекает, но с большой энергией активации (около 250 кДж/моль).
5.3.2. Запрет по орбитальному моменту (второе правило Вигнера – Витмера)
В ходе химического превращения сохраняется орбитальный момент системы, т.е. реакция запрещена по орбитальному моменту, если среди возможных орбитальных состояний продуктов реакции нет состояния, совпадающего с одним из орбитальных состояний исходных веществ. Это правило применимо для реакций, в которых сохраняется осевая (аксиальная) симметрия, т.е. атомы реагирующих молекул все время находятся на одной линии и могут образовывать только линейные молекулы.
Легко показать, что в приведенных выше примерах реакций образования О2 и ОН из атомов орбитальные моменты в реакциях сохраняются. Действительно, в первой реакции имеем для исходных веществ орбитальные моменты λ1 = λ2 = 1. Следовательно, система имеет состояния с Λ = 2, 1 и 0. Продукт реакции находится в состоянии ΣΛ = 0. Так как это состояние имеется среди состояний исходных веществ, то реакция разрешена по орбитальному моменту.
Для удобства анализа реакций можно воспользоваться таблицами, построенными с помощью теории групп. Табл. 3 позволяет найти энергетические состояния линейной системы, состоящей из двух линейных молекул. Для этого необходимо найти столбец, соответствующий состоянию одной молекулы, затем – строку, соответствующий состоянию другой молекулы, на пересечении находится набор состояний, характеризующий систему. Если реагируют или получаются атомы, то их состояния, отвечающие симметрии относительно оси симметрии, можно определить с помощью табл. 4.
Рассмотрим, например, реакцию
N2O (1Σ) + H (2S) → N2 (1Σ) + OH (2Π).
Спин в этой реакции сохраняется, а орбитальный момент – нет. Типу симметрии атомного состояния атома H (2S) соответствует (см. табл. 4) тип симметрии относительно воображаемой оси связи Σ. Суммарное состояние исходных веществ (см. табл. 3) Σ + Σ = Σ, а продуктов Σ + Π = Π. Так как состояния исходных веществ и продуктов не совпадают, то реакция «запрещена». На самом деле эта реакция идет, но с определенной энергией активации порядка 50 – 90 кДж/моль. Изменить симметрию можно за счет различных внешних воздействий, приводящих к изменению
5
состояния. Чаще всего желаемый эффект достигается воздействием на исследуемые молекулы фотонов, электронно возбужденных атомов или катализаторов.
Таблица 3. Результирующая симметрия волновой функции линейной системы, состоящей из двух линейных молекул
Первая |
|
Вторая молекула |
Первая |
|
Вторая молекула |
|
||
молекула |
|
молекула |
|
|
||||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
Σ+ |
Σ− |
Π |
Π |
Π |
Π |
Σ+, Σ−, |
Π, Φ |
Σ+ |
Σ+ |
Σ− |
Π |
|
|
|
|
|
Σ− |
Σ− |
Σ+ |
Π |
|
|
|
Π, Φ |
Σ+, Σ−, Γ |
Таблица 4. Симметрия состояний атома относительно оси симметрии
Тип симметрии |
Тип симметрии |
Тип симметрии |
Тип симметрии |
|
относительно оси |
относительно оси |
|||
атомного состояния |
атомного состояния |
|||
симметрии |
симметрии |
|||
|
|
|||
|
|
|
|
|
Sg |
Σ+ |
Pu |
Σ+ Π |
|
Su |
Σ− |
Dg |
Σ+ Π |
|
Pg |
Σ− Π |
Du |
Σ− Π |
Например, разложение N2O при фотосенсибилизации ртутью (участвуют атомы ртути в
возбужденном состоянии)
N2O (1Σ) + Hg (2P) → N2 (1Σ) + O (3P) + Hg (1S)
протекает без осложнений, поскольку не ограничивается спиновыми и орбитальными запретами. К сожалению, класс реакций, подчиняющихся правилам запрета Вигнера – Витмера, очень
ограничен, хотя многие из них важны, например, реакции в пламени, при взрывах и в фотохимии. Правила оказались неприменимы при рассмотрении многоатомных нелинейных молекул.
5.3.3. Правило сохранения орбитальной симметрии Вудворда – Хоффмана
Существуют реакции, протекающие с высоким потенциальным барьером, которые нельзя описать с помощью правил Вигнера – Витмера. Реакция димеризации этилена в циклобутан
C2H4 + C2H4 → C4H8
является примером превращения в нелинейной системе, имеющей большую энергию активации, наличие которой не может быть предсказано правилами Вигнера – Витмера.
Вудворд и Хоффман показали, что этот барьер может быть связан с несохранением симметрии определенных молекулярных орбиталей в ходе реакции. Если занятые орбитали исходных веществ одинаковы по симметрии только с занятыми орбиталями продуктов реакции, то реакция по правилу Вудворда – Хоффмана считается разрешенной и протекает практически без потенциального барьера. Если же какая-либо из занятых орбиталей реагирующих веществ коррелирует с незанятыми орбиталями продуктов реакции (или наоборот), то реакция является запрещенной. При этом она может протекать, но обычно с высоким потенциальным барьером или совсем по другому механизму.
При анализе сохранения признаков симметрии, особенно в сложных случаях, пользуются
корреляционными диаграммами (КД). Различают КД орбиталей и КД состояний. КД орбиталей –
это схема, которая в координатах «энергия» – « координата реакции» показывает переход орбиталей исходных веществ в орбитали продуктов реакции той же симметрии. На КД состояний изображаются уровни энергий всей системы в целом и их изменение в ходе реакции. При построении КД необходимо соблюдать два правила: правило сохранения симметрии волновой функции системы в ходе реакции (спин S, орбитальный момент λ и Λ, симметрия g и u, + и –) и
6
правило непересечения, согласно которому корреляционные линии, отвечающие состояниям (орбиталям) одинаковой симметрии, не пересекаются. Если при построении КД такое пересечение выявляется, то это означает запрет на протекание реакции, т.е. реакция протекает с высоким потенциальным барьером.
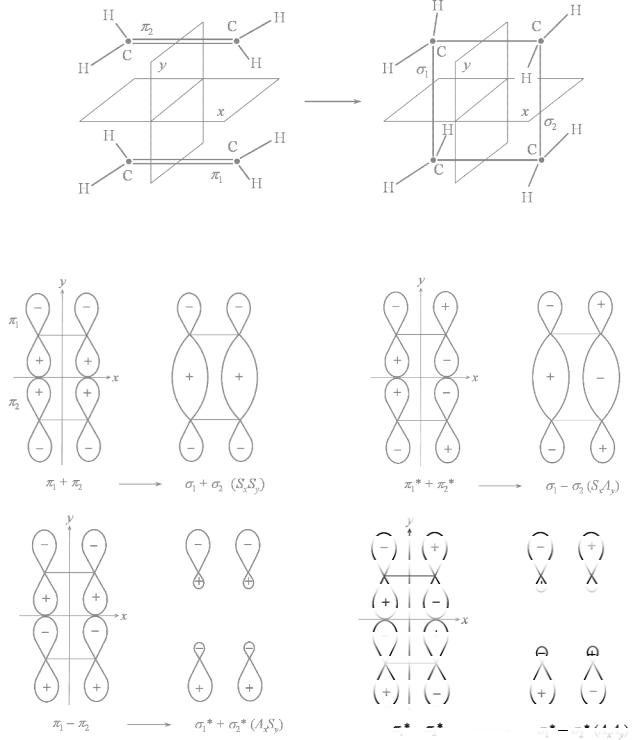
Рассмотрим КД орбиталей для реакции димеризации этилена в циклобутан (рис. 21). Все МО будем классифицировать как симметричные (S) и антисимметричные (A) относительно плоскостей x и y, которые пересекают рвущиеся и образующиеся связи.
Рис. 21. Плоскости симметрии, относительно которых классифицируются МО двух молекул этилена и циклобутана в реакции 2C2H4 → C4H8.
Рис. 22. Орбитали взаимодействующих молекул этилена и циклобутана. В скобках указаны свойства симметрии относительно плоскостей x и y на рис. 21.
Сущность рассматриваемой реакции с точки зрения локализованных МО состоит в том, что π1- и π2-орбитали молекул этилена превращаются в σ1- и σ2-орбитали циклобутана. Поскольку
7

замена двукратно занятых МО их линейными комбинациями не меняет волновой функции (свойство детерминанта Слэтера), для удобства заменим π1- и π2-МО молекул этилена их линейными комбинациями π1 + π2 и π1 – π2. Последние обладают соответствующими свойствами симметрии относительно плоскости x. По этим же причинам заменим σ1- и σ2-МО циклобутана на их линейные комбинации σ1 + σ2 и σ1 – σ2. Эти комбинации, а также соответствующие возбужденные МО (с индексом *) показаны на рис. 22; здесь же указана их симметрия относительно плоскостей x и y (Sx, Sy, Ax, Ay).
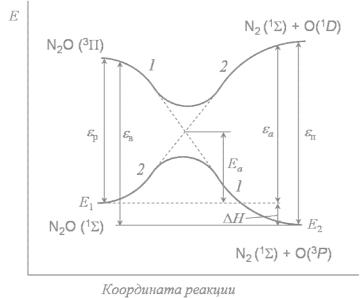
Исходя из схем рис. 22, построим КД орбиталей (рис. 23). На ней прямыми линиями
соединены МО одинаковой симметрии. Главный |
вывод из |
нее |
состоит |
в |
том, |
что |
занятая |
|||
|
(связывающая) |
орбиталь |
системы |
|||||||
|
исходных веществ |
π1 |
– |
π2 |
симметрии |
|||||
|
AxSy |
превращается |
|
(говорят |
– |
|||||
|
коррелирует) |
в |
|
|
незаполненную |
|||||
|
(разрыхляющую) МО продукта σ1* + |
|||||||||
|
σ2* той же симметрии; в то же время |
|||||||||
|
занятая (связывающая) σ1 – |
σ2-орбиталь |
||||||||
|
продукта реакции симметрии SxAy |
|||||||||
|
получается |
|
из |
|
|
незанятой |
||||
|
(разрыхляющей) |
|
МО |
исходного |
||||||
|
вещества π1* + π2* той же симметрии. |
|||||||||
|
Как видно, верхняя занятая МО |
|||||||||
|
исходных |
веществ |
π1 |
– |
π2 |
имеет |
||||
|
симметрию AxSy, а соответствующая МО |
|||||||||
|
продуктов |
σ1 |
– |
σ2 |
|
имеет |
другую |
|||
|
симметрию |
SxAy, |
|
т.е. |
они |
не |
||||
|
коррелируют между собой. Этот факт в |
|||||||||
Рис |
соответствии с |
правилом |
Вудворда |
– |
||||||
. 23. Корреляционная диаграмма молекулярных |
Хоффмана указывает на то, |
|
|
|
||||||
орбиталей для реакции 2C2H4 → C4H8. |
что реакция |
|||||||||
|
должна |
протекать |
|
с |
высоким |
|||||
потенциальным барьером. И действительно, энергия активации такого превращения так велика, что реакция практически не идет. Однако эта же реакция легко осуществляется после возбуждения молекул светом.
На рис. 24 показана КД состояний той же реакции. Состояние определяется набором МО с указанием их заполнения электронами. Отличие этой диаграммы от предыдущей состоит в том, что вместо уровней МО здесь взяты уровни энергии системы в
целом. |
Симметрия |
состояния |
определяется с учетом |
заполнения |
|
уровней электронами по правилу: если два электрона находятся на двух уровнях с одинаковой симметрией S или A, то результирующая симметрия двух электронов будет S (S × S = S, A ×
A = S), а если они на уровнях различной симметрии, то S × A = A. Легко
заметить, что при двухэлектронном заполнении орбиталей состояние всегда
будет S. Поэтому КД состояний строят по КД орбиталей способом, который виден из сравнения рис. 23 и 24.
8
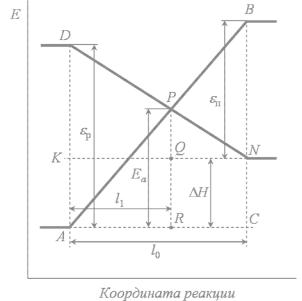
На КД состояний энергетические уровни для заполненных и незаполненных МО пересекаются. Положение точки пересечения может быть использовано для приближенной количественной оценки потенциального барьера (энергии активации). Рассмотрим в качестве примера реакцию распада молекулы N2O, которая имеет довольно высокий потенциальный барьер, но все же осуществима. Молекула оксида азота (I) сравнительно устойчивое соединение. Разложение на N2 и O начинается только при 1100 – 1200 К. В основном состоянии разрыв связей N–O и N–N при термическом инициировании не может осуществиться из-за запрета по спину (см.
раздел 5.3.1). На рис. 25 приведена энергетическая схема возможного пути протекания реакции
N2O (1Σ) → N2 (1Σ) + O (3P).
Переход в продукты может быть осуществлен путем предварительного перевода N2O (1Σ) в другое (возбужденное) энергетическое состояние N2O (3П). При этом затрачивается энергия εp и
снимается запрет по спину на протекание реакции (рис. 25, кривая 1) N2O (1Σ) → N2O (3П) → N2 (1Σ) + O (3P).
Второй путь протекания этой реакции возможен (см. рис. 25, кривая 2), когда в результате реакции
получается кислород в возбужденном состоянии:
N2O (1Σ) → N2 (1Σ) + O (1D).
Этот переход разрешен по спину и орбитальной симметрии (см. табл. 3 и 4). Выделение энергии
приводит к переходу реакции в нормальное состояние N2 (1Σ) и O (3P).
Получим выражение для энергии активации Ea через энергетические параметры исходных веществ и продуктов реакции (рис. 26). Из подобия треугольников APR и ABC следует
|
|
|
Ea |
|
= |
l1 |
, |
|
|
|
|
|
εп + |
|
|
|
|
|
|||
|
|
H l0 |
|
|
|
|
||||
или |
|
|
|
|
|
|
|
|
|
|
|
l1 |
|
(ε + |
H ) = E |
a |
. |
(105) |
|||
|
l0 |
|
||||||||
|
|
п |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Рис. 25. Энергетическая схема возможных путей протекания реакции N2O → N2 + O.
Из подобия треугольников NPQ и NDK получаем
|
Ea − H |
= |
l0 − l1 |
= 1 − |
l1 |
, |
||
|
ε p − H |
l0 |
l0 |
|||||
|
|
|
|
|||||
или |
|
|
|
|
|
|||
|
|
l1 |
(εp − |
H )= εp − Ea . |
(106) |
|||
|
|
|
||||||
|
|
l0 |
|
|
|
|
|
|
Для решения системы уравнений (105) и (106) с двумя неизвестными l1/l0 и Ea сначала сложим их:
l |
= |
|
|
ε p |
|
|
1 |
|
|
|
. |
(107) |
|
l |
ε |
p |
+ ε |
|||
0 |
|
|
п |
|
||
Подставляя значение l1/l0 в уравнение (105), получаем искомое выражение для энергии активации
Ea: |
|
|
ε p |
|
|
|
|
Ea |
= |
|
|
(εп |
+ H ). |
(108) |
|
εp |
+ ε |
|
|||||
|
|
п |
|
|
|||
Энергии возбуждения исходных веществ εp и продуктов реакции εп обычно известны. Уравнение
(108)справедливо и для экзотермических реакций.
Оценим, например, энергию активации для реакции термического распада молекулы N2O.
Для этой реакции известны энергии возбуждения εp = 394 кДж/моль, εп = 193 кДж/моль и энтальпия реакции H = 168 кДж/моль. Энергия активации, рассчитанная по формуле (108), равна 252 кДж/моль, что хорошо согласуется с экспериментальным значением Ea.
9
Эмпирически установленное правило Вудворда – Хоффмана было теоретически подтверждено впоследствии квантово- химическими расчетами по методу МО и проанализировано с помощью аппарата теории групп. При этом точкам пересечения линий на КД были сопоставлены МО переходного состояния. Положение переходного состояния относительно реагентов и продуктов согласуется с предсказаниями постулата Хэммонда: экзотермической реакции соответствует реагентоподобное переходное состояние.
В настоящее время квантово-химический анализ реакционной способности, использующий КД, широко применяется для предсказания синтезов органических соединений, для анализа разнообразных газофазных реакций, реакций в конденсированных средах, а также элементарных стадий каталитических процессов.
Рис. 26. Схематическое изображение корреляционной диаграммы с пересечением энергетических уровней.
5.4. Индексы реакционной способности
Индексами реакционной способности (ИРС) называются полученные в результате квантово- химических расчетов электронные и энергетические характеристики системы, которые коррелируют с экспериментальными данными о реакционной способности. Из этого общего определения ясно, что таких индексов существует достаточно много и с каждым годом становится все больше. Практическое применение ИРС следует рассматривать в плане образования набора расчетных величин – дескрипторов, коррелирующих с конкретными свойствами молекулярной системы.
В современной квантовой химии значимость метода ИРС определяется его применением для изучения свойств больших молекул и твердых тел. Использование метода ИРС для биологически активных молекул привело к формулировке Ханшем задачи QSAR (Quantitative Structure-Activity Relationship). Эта задача состоит в создании моделей на основе количественных корреляций между свойствами (например, биологической активностью) больших молекул и суммой химических дескрипторов составляющих их функциональных групп. Предполагается, что любое свойство большой молекулы приближенно определяется суммой свойств входящих в нее функциональных групп. Наибольшее внимание сейчас привлекают обратные QSAR-задачи, состоящие в генерации новых структур с заданными свойствами – молекулярный дизайн.
Рассмотрим наиболее распространенные ИРС. Они делятся на две группы. Первые определяются в приближении реагирующей молекулы (приближении локализации), когда учитывают свойства переходного состояния химической реакции, вторые – в приближении изолированной молекулы с учетом статических свойств лишь исходных веществ: структуры и энергии граничных МО, зарядов, порядков связей и т.д.
5.4.1. Молекулярный электростатический потенциал
Распространенным и информативным статическим ИРС молекулы является молекулярный электростатический потенциал (МЭП). Его физический смысл следующий. Пусть заряд q1 создает в точке пространства с радиус-вектором r МЭП U(r). Если в эту точку поместить точечный заряд q, то энергия электростатического взаимодействия между зарядом q и зарядом q1 будет равна qU. Электронная плотность ρ(r), рассчитываемая с использованием электронной волновой функции, ищется в виде детерминанта Слэтера, построенного из одноэлектронных волновых функций – МО. Каждая МО Ψi есть линейная комбинация базисных функций ψν с
10
коэффициентами разложения ciν (см. уравнение (79)). В каждой точке r пространства внутри и вне молекулы МЭП имеет вид:
U (r ) = −∫ |
ρ (r ) |
|
|
Z |
|
e |
|
|
|
|
|
|
|
|
|
||||||||
|
dUi |
+ ∑ |
|
α |
|
|
|
|
, |
(109) |
|
|
|
|
|||||||||
r − r |
|
r − |
|
R |
|
|
|||||
|
i |
|
α |
|
|
α |
|
|
|||
где Zα e – заряд ядра α; Rα – радиус-вектор ядра α.
Особый интерес МЭП представляет потому, что необходимая для его расчета электронная плотность ρ(r) может быть не только рассчитана теоретически, но и получена из эксперимента, например, из рентгеноструктурного анализа.
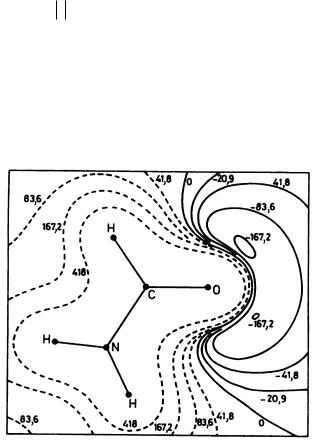
Лучший способ визуализации МЭП состоит в построении контурных карт изопотенциалов, создаваемых молекулами. В качестве примера на рис. 27 карта МЭП формамида. Глубокие хорошо локализованные потенциальные минимумы показывают, что молекула формамида будет протонироваться по неподеленным электронным sp2-парам атома кислорода.
Карты МЭП часто позволяют интерпретировать электрофильные и нуклеофильные свойства молекул в основном и электронно-возбужденном состояниях. Они чувствительны даже к малым изменениям молекулярной структуры. В последние годы это привело к использованию МЭП в качестве дескрипторов реакционной и селективной способности в QSAR-задачах. При этом только один из реагентов рассматривается в явном виде, а второй заменяется точечным зарядом.
Для существенно ионных соединений эффективные заряды на атомах в приближении «первого порядка» дают МЭП, пригодный для предсказания направления атаки при взаимодействии. МЭП позволяет оценить только электростатическую составляющую энергии межмолекулярных взаимодействий. Но для корректного определения последней не меньшее значение может иметь сумма индукционной (поляризационной) и дисперсионной составляющих.
Оценить эту энергию позволяет рассмотрение взаимодействия граничных МО реагирующих молекул (см. раздел 5.5).
5.4.2. Валентность атомов и порядок связи
Реакционную способность молекул с давних пор было принято характеризовать такими статическими ИРС, как валентность атомов и кратность связей. В квантовой химии соответствующие индексы валентности VA, порядка связи по Коулсону РAB или по Вайбергу IAB
определяются структурой МО μ и ν атомов А и В, точнее произведениями диагональных и недиагональных элементов матрицы зарядов и порядков связей Рμν и матрицы интегралов перекрывания Sμν:
PAB = ∑Pμν Sμν , |
μ A , ν B ; |
(110) |
|||||
μ ,ν |
|
|
|
|
|
||
IAB = ∑ |
|
Pμν Sμν |
|
2 |
, |
μ A , ν B ; |
(111) |
|
|
||||||
μ ,ν |
|
VA = ∑PAB . |
(112) |
||||
|
|
||||||
|
|
|
|
|
A¹B |
|
|
Индексы связей сохраняют свои значения при отклонении длины связи от равновесной. Например, индексы связей С– С в молекулах этана, этилена и ацетилена при варьировании
11

равновесной длины связи от ординарной до тройной сохраняют близость к значениям соответственно 1,0; 2,0; 3,0 (табл. 5).
Таблица 5. Индексы Вайберга IСС, вычисленные при межатомных расстояниях, равных равновесным длинам связей RCC в молекулах этана, этилена и ацетилена.
Молекула |
RCC = 1,52 Å |
RCC = 1,34 Å |
RCC = 1,20 Å |
H3C–CH 3 |
0,99 |
1,03 |
1,07 |
H2C=CH2 |
1,95 |
1,99 |
2,02 |
HC≡CH |
2,91 |
2,94 |
2,96 |
|
|
|
|
5.4.3. Энергия диссоциации химической связи в молекулярной системе
Недавно при анализе колебаний молекул был предложен новый ИРС – энергия активации реакции разрыва химической связи. При феноменологическом изучении взаимодействий молекул с поверхностью металла оказалось, что потенциал взаимодействия хорошо аппроксимируется потенциалом Морзе (11) с параметрами DAB и αAB
При использовании эффективного оператора энергии ангармонической колебательной задачи был введен индекс реакционной способности D, который для каждой валентной связи в молекулярной системе рассчитывается как параметр DAB потенциала Морзе с поправкой на энергию нулевых колебаний:
|
|
|
|
D = DAB – |
Eнк. |
|
(113) |
|
Теоретические и экспериментальные оценки энергии связи C–Y |
в молекулах |
вида YCX3 |
||||||
представлены в табл. 6. Они находятся в весьма удовлетворительном согласии. |
|
|
||||||
Таблица 6. Индекс реакционной способности D на основе потенциала Морзе и |
||||||||
экспериментальные значения Dэ энергии связей C–Y в молекулах YCX3. |
|
|
||||||
|
Параметры* |
DCCl3 |
HCCl3 |
HCF3 |
ClCF3 |
ICF3 |
|
|
|
|
|
|
|
|
|
|
|
|
DAB, см-1 |
32437,2 |
30675,3 |
38660,0 |
30500,0 |
19393,4 |
|
|
|
|
Eнк, см-1 |
1137,4 |
1487,3 |
1535,1 |
267,1 |
166,8 |
|
|
D, кДж/моль |
374,5 |
349,2 |
444,2 |
361,7 |
230,0 |
|
|
|
Dэ, кДж/моль |
369,2 |
358,2 |
446,4 |
360,2 |
231,5 |
|
|
|
|
|
|
|
||||
|
* DAB – |
параметр потенциала Морзе (энергия диссоциации); |
Енк – энергия нулевых колебаний осциллятора С–H |
|||||
|
(C–D). 1 |
см–1 = 1,1964807·10-2 кДж/моль. |
|
|
|
|
|
|
В случае связи, локализованной в квантово-химическом смысле, при диссоциации по которой симметрия системы сохраняется, индекс реакционной способности D совпадает по величине с энергией активации разрыва этой связи и предоставляет возможность оценки последней из колебательного спектра молекулы.
5.5. Теория граничных орбиталей
Оценки реакционной способности особенно просты, если использовать теорию возмущений. В распространенном варианте теории возмущений энергия стабилизации представляется в виде суммы вкладов от взаимодействия между МО реагентов. Наибольший вклад в сумму дают, как правило, взаимодействия так называемых граничных орбиталей, в качестве которых рассматривают высшую занятую МО (ВЗМО) и низшую свободную МО (НСМО). В радикалах или возбужденных молекулах роль одной из этих орбиталей или их обеих могут играть однократно занятые МО. Наиболее важная характеристика граничных МО – их парциальная электронная
12
плотность, т.е. плотность на отдельных атомах, называемая граничной электронной плотностью. Граничная электронная плотность определяет предпочтительные места атаки.
Основной постулат теории: реакции легче всего протекают в случае максимального перекрывания (смешивания) граничных МО, которые, как правило, вносят наибольший вклад в энергию взаимодействия реагентов. Перекрывание приводит к переносу заряда с ВЗМО донора на НСМО акцептора. При этом положение с наибольшей граничной плотностью НСМО, как правило, наиболее благоприятно для нуклеофильной атаки; электрофильная атака протекает преимущественно по положению, где граничная плотность ВЗМО наибольшая. Взаимодействие с переносом заряда между ВЗМО одного реагента и НСМО другого приводит к возрастанию граничной плотности в области перекрывания.
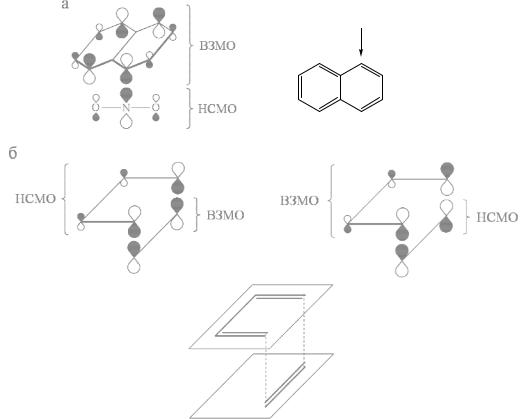
Для описания реакционной способности необходимо знать только вид граничных МО реагирующих молекул, который, как правило, определяется при помощи простейших квантово- химических расчетов методами МО. Для выявления оптимального пути реакции необходимо, зная структуру граничных МО, выделить такой способ сближения реагентов, который соответствовал бы максимальной величине интеграла перекрывания между данными МО, а, следовательно, их наибольшему смешиванию в промежуточном комплексе и наибольшему переносу заряда с ВЗМО одной молекулы на НСМО другой. Например, вид ВЗМО нафталина и НСМО иона нитрония, имеющих максимальную плотность в соположении нафталина и на атоме N иона нитрония соответственно, объясняет, почему нитрование нафталина происходит в основном в α-положение (рис. 28а). Вид граничных МО бутадиена и этилена, имеющих одинаковую симметрию, объясняет предпочтительность супра-супраповерхностного способа их сближения в диеновом синтезе (рис. 28б).
NO2+
Рис. 28. Орбитальные взаимодействия при нитровании нафталина (а) и в диеновом синтезе (б).
В ряде случаев невозможно четко выделить граничные орбитали, поскольку контролировать реакцию могут несколько (или даже целые полосы) высоколежащих занятых и низколежащих свободных МО. Так, протонирование пиридина происходит не в положение с максимальной амплитудой высшей занятой π-орбитали, а по неподеленной электронной паре атома N.
13