

Нарушение обмена аминокислот
|
Нарушения обмена фенилаланина
|
Схема 29 |
|
1.Фенилкетонурия1 типа (классическая) |
|
|
2.Фенилкетонурия 2 типа. Тетрагидробиоптерин-дефицитная гиперфенилаланинемия. |
Схема 30 Схема 31 Схема 32 |
|
а) Недостаточность дигидробиоптерин редуктазы. |
|
|
б) Недостаточность6‑пирувоилтетрагидроптерин синтазы. |
|
|
в). Недостаточность гуанозинтрифосфат-циклогидролазы-1.
ДОФА-чувствительная дистония(болезнь Сегавы) |
|
|
3. Материнская ФКУ
|
|
|
Нарушение обмена тирозина. Тирозинемии |
Схема 33 |
|
Тирозинемия 1 типа
|
|
|
Тирозинемия 2 типа
|
|
|
Тирозинемия новорожденных
|
|
|
Алкаптонурия
|
|
|
Альбинизм (ахроматоз). |
|
|
Паркинсонизм
|
|
|
Нарушение обмена метионина и цистеина |
Схема 34 |
|
Гомоцистеинемия |
|
|
Цистиноз - это накопление цистина |
Схема 35 Схема 36 Схема 37 Схема 38 |
|
Нарушение распада лейцина, валина, изолейцина |
|
|
Лейциноз (болезнь кленового сиропа, БКС). |
Схема 39 |
|
Изовалератацидемия |
Схема 40 |
|
Нарушение обмена триптофана |
Схема 41 |
Top of Form
Нарушения обмена фенилаланина
Фенилаланин относится к незаменимым аминокислотам, поскольку ткани животных не обладают способностью синтезировать его бензольное кольцо. При любых нарушениях превращения его в тирозин развиваетсяфенилкетонурия.
Выделяют несколько типов фенилкетонурии:
классическая(1 типа),
тетрагидробиоптерин-дефицитная гиперфенилаланинемия(2 типа),
материнская (3 типа).
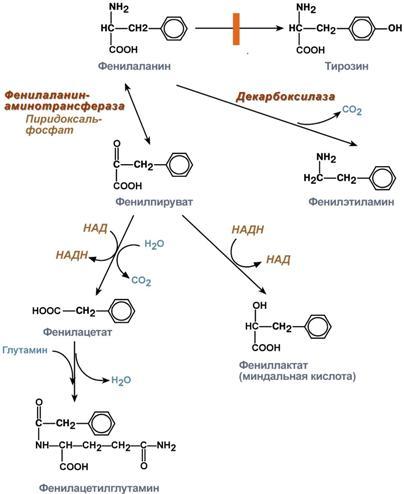
Превращение фенилаланина при фенилкетонурии
1. Фенилкетонурия 1 типа (классическая)
Фенилкетонурия 1 типа является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10 000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100 000 в Японии.
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин гидроксилазы(фенилаланин-4-монооксигеназы), обеспечивающей превращение фенилаланина в тирозин. Ребёнок должен получить две повреждённые аллели от обоих родителей – только в этом случае будет проявление болезни. Фермент имеется только в печени, почках, поджелудочной железе. В настоящее время известно более 400 мутаций этого фермента ведущие к заболеванию.
Патогенез
В патогенезе ФКУ имеют значение многие обстоятельства, в частности:
значительное накопление в тканяхи жидкостях больного организма фенилаланина и его производных (фенилпировиноградная,фенилмолочная (миндальная),фенилуксусная, гиппуровая кислоты,фенилэтиламин,фенилацетилглютамин) и вызванный имиацидоз (В клинической практике пробой на синтез пара-аминогиппуровой кислоты (проба Квика-Пытеля) устанавливают способность печени обезвреживать ядовитые вещества. (hippos, - horse);
прямое токсическое действиеуказанных веществ на центральную нервную систему, которое заключается вторможениифенилаланиномактивности рядаферментов,в том числепируваткиназы(гликолиз),тирозиназы(синтез меланина),тирозин-гидроксилазы(синтез катехоламинов);
нарушение синтеза серотонина, т.к. фенилаланин-4-монооксигеназа одновременно осуществляет гидроксилирование триптофана до5-гидрокситриптофана, предшественника серотонина,
конкурентное снижениефенилаланиномтранспортав клетки ароматических аминокислот – триптофана и тирозина,
нарушение синтезапростых и сложныхбелковв тканях, что вызывает тяжелые повреждения мозга и нарушение функции печени у большинства больных.