

1Foundation of Mathematical Biology / Foundation of Mathematical Biology
.pdfUCSF
Lecture III: Resampling and permutation-based methods
Resampling methods
♦Efron’s bootstrap and related methods
♦Resample with replacement from a population distribution constructed from the empirical discrete observed frequency distribution
Permutation-based methods
♦Shatter the relationship on which your statistic is computed
♦Empirical method to derive the null distribution of a particular statistic given the precise context in which it is applied
We will focus on hypothesis testing and will address the problem of multiple comparisons
UCSF |
Two basic cases |
|
|
|
|
Unpaired data, multiple classes
We have computed some statistic based on the class assignments
The idea is to empirically generate the null distribution of this statistic by repeatedly randomly permuting the class membership
Paired data, or, more generally, vectorial data of any dimension
Each sample has a vector
We compute a statistic that depends on the relationship of variables within the vectors over all samples
The idea is to empirically generate the null distribution of this statistic by repeatedly randomly permuting parts of the vectors across the samples
UCSF |
Unpaired data: |
|
Rewrite our statistical function to take values+classes |
||
|
|
|
X1...X n1
Y1...Yn2
Z1...Zn3
f ( X* ,Y*, Z*)
V1...Vn1 +n2 +n3 = X* ,Y*, Z* C1...Cn1 +n2 +n3 = class
*
f ' (V*, C)
We need the distribution of f’ under either of the following
♦Random permutations of the order of the vector V
♦Random resamplings of V (with replacement) from V itself (this is equivalent to the bootstrap procedure that Jane described)
Note: We have essentially converted this to a paired data set
UCSF |
What is the intuition? |
|
|
|
|
Each permutation or resampling is a simulated experiment where we know the null hypothesis is true
By generating the empirical distribution of f’ under many random iterations, we get an accurate picture of the likelihood of observing a statistic of any magnitude given the exact distributional and size characteristics of our samples
To assess significance of a statistic of value Z
♦Perform N permutations as described
♦Compute f’ for each
♦Count the number that meet or exceed Z (= nbetter)
♦Significance = nbetter/N (the probability that we will observe a statistic as good as Z under the null hypothesis)
UCSF |
Example: Alternative to T test |
|
|
|
|
We sample from the standard normal distribution to get
X1…X10 and Y1…Y10
Using the T test with equal variances, we compute the following from our sample:
The critical value for this test given 18 DF is 2.101
If we do this 100,000 times to check how accurate the critical value is, we get a proportion of
0.0509 t scores that exceed this critical value
t =
X −Y
S 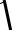 1n + m1
1n + m1
S = |
(n −1)U + (m −1)V |
|
n + m − 2 |
So, statistical theory works fine. How does permutation?

UCSF
The permutation approach yields similar results
Permutation simulation
We sample from the standard normal distribution to get
X1…X10 and Y1…Y10
We compute the T test with equal variances
For each sample, we perform 1000 permutations, recompute t, and count the number better than our initial t.
What proportion of instances yield p values better than 0.05?
With 10,000 simulations, 0.0483
As with the critical value from statistical theory, we get an appropriate proportion by direct simulation
If we resample with replacement, we get 0.0492
UCSF |
We don’t have to use a “real statistic” |
|
|
|
|
We sample from the standard normal distribution to get
X1…X10 and Y1…Y10
We compute the absolute difference of means
For each sample, we perform 1000 permutations, recompute, and count the number better than our initial difference.
What proportion of instances yield p values better than 0.05?
With 10,000 simulations, 0.0483
This is exactly the same as before. We did not have to normalize by the pooled variance.
UCSF |
Suppose our statistical assumptions fail? |
|
|
|
|
We sample from the standard normal distribution to get
X1…X10
We sample from a normal distribution with mean 0, and variance 16 to get Y1…Y10
So, we don’t have equal variances
What proportion of instances yield p values better than 0.05?
♦T-test: 0.061
♦Permutation: 0.053
When our statistical assumptions fail, permutationbased methods give us a better estimate of significance

UCSF
Paired data: Permute on the data pairing
X1...X n |
X1...X n |
Y1...Yn |
Y1...Yn |
f ( X ,Y ) |
f ' ( X |
,Y ) |
* * |
* |
* |
We need the distribution of f’ under either of the following
♦Random permutations of the order of the vector Y
♦Random resamplings of X and Y (with replacement) from X and Y themselves (like the bootstrap procedure)
Note that each of the Xi or Yi can be vectors themselves.

UCSF
Expression array example: Lymphoblastic versus myeloid leukemia
Lander data
♦6817 unique genes
♦Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia (ALL and AML) samples
♦RNA quantified by Affymax oligo-technology
♦38 training cases (27 ALL, 11 AML)
♦34 testing cases (20/14)
We will consider whether any of the genes are differently expressed between the ALL and
AML classes
R E P O R T S
Molecular Classification of
Cancer: Class Discovery and
Class Prediction by Gene
Expression Monitoring
T.R. Golub,1,2*† D. K. Slonim,1† P. Tamayo,1 C. Huard,1 M. Gaasenbeek,1 J. P. Mesirov,1 H. Coller,1 M. L. Loh,2
J. R. Downing,3 M. A. Caligiuri,4 C. D. Bloomfield,4
E.S. Lander1,5*
Although cancer classification has improved over the past 30 years, there has been no general approach for identifying new cancer classes (class discovery) or for assigning tumors to known classes (class prediction). Here, a generic approach to cancer classification based on gene expression monitoring by DNA microarrays is described and applied to human acute leukemias as a test case. A class discovery procedure automatically discovered the distinction between acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) without previous knowledge of these classes. An automatically derived class predictor was able to determine the class of new leukemia cases. The results demonstrate the feasibility of cancer classification based solely on gene expression monitoring and suggest a general strategy for discovering and predicting cancer classes for other types of cancer, independent of previous biological knowledge.
SCIENCE VOL 286 15 OCTOBER 1999