

Computational Methods for Protein Structure Prediction & Modeling V1 - Xu Xu and Liang
.pdf10 Homology-Based Modeling of Protein Structure
Zhexin Xiang
10.1 Introduction
10.1.1Structural Genomics and Homology Modeling
The human genome project has already discovered millions of proteins (http://www.swissprot.com). The potential of the genome project can only be fully realized once we can assign, understand, manipulate, and predict the function of these new proteins (Sanchez and Sali, 1997; Frishman et al., 2000; Domingues et al., 2000). Predicting protein function generally requires knowledge of protein three-dimensional structure (Blundell et al., 1978; Weber, 1990), which is ultimately determined by protein sequence (Anfinsen, 1973). Protein structure determination using experimental methods such as X-ray crystallography or NMR spectroscopy is very time consuming (Johnson et al. 1994). To date, fewer than 2% of the known proteins have had their structures solved experimentally. In 2004, more than half a million new proteins were sequenced that almost doubled the efforts in the previous year, but only 5300 structures were solved. Although the rate of experimental structure determination will continue to increase, the number of newly discovered sequences grows much faster than the number of structures solved (see Fig. 10.1).
Fortunately, many protein sequences are evolutionarily related, and thus can be classified into different families. Proteins in the same families frequently have noticeable similarities and thus share three-dimensional architecture, which allows a structural description of all proteins in a family even when only the structure of a single member is known. This evolutionary relationship provides the rationale for structural genomics, a systematic and large-scale effort toward structural characterization of all proteins, where a representative protein in each family is chosen to be solved experimentally with the rest reliably predicted by a homology modeling method (Goldsmith-Fischman and Honig, 2003; Al-Lazikani et al., 2001a). Fold recognition has also become an important tool that supplements sequence-based methods to detect remote homologues. However, the line between traditional homology modeling and fold recognition has diminished due to the progress in the alignment sensitivity and the increase in database size. Ab initio structure methods have made notable progress in recent years and are extremely important, not only for what they can accomplish but also for what they can teach us about protein folding (Bonneau and Baker, 2001). The progress in ab initio prediction makes it possible in a few cases to refine homology models to the accuracy of low-resolution X-ray
319

320 |
Zhexin Xiang |
Fig. 10.1 Number of protein sequences and structures available each year. Blue bar denotes the number of protein sequences in SWISS-PROT, red bar is the number of protein structures in PDB.
structures. In fact, if we assume that a native protein structure is at the global freeenergy minimum, comparative modeling is a simple scheme to focus the search of conformation space by minimally disturbing those existing solutions, i.e., the experimentally solved structures. The obvious advantage is that the comparative modeling technique relaxes the stringent requirements of force field accuracy and prohibitive conformational searching, because it dispenses with the calculation of a physical chemistry force field and replaces it, in large part, with the counting of identical residues between template and target sequences.
Currently there are about 2 million protein sequences in Swiss-Prot and TrEMBL, but only 7677 protein families have been identified according to the Pfam database ( http://pfam.wustl.edu/). This number is strongly dependent on the sequence similarity cutoffs used to cluster the sequence space. If 30% sequence identity cutoff is used, which is generally considered as a threshold for successful homology modeling, statistical estimates place the number somewhere between 10,000 and 30,000 for all proteins in Nature (Liu et al., 2004), but only a fraction of which have distinct spatial arrangements (Brenner et al., 1997). Ninety percent of protein structures deposited today share a similar fold to others already in the PDB. Many of these solved structures are site-directed mutants or inhibitor-bound complexes of previously deposited proteins, and many are related members of families of proteins with similar sequences and closely related three-dimensional structures. Even proteins with no sequence homology can have very similar folds; for example, the sulfate and phosphate binding proteins, the transferrins, and the porphobilinogen deaminase have similar bilobal anion binding structures but not significant sequence identities. Protein topologies such as the four -helix bundle, the -nucleotide binding motif,
10. Homology-Based Modeling of Protein Structure |
321 |
the -jelly roll, the -barrel, and the -immunoglobulin domain have been found in a wide range of protein structures (Johnson et al., 1994; Al-Lazikani et al., 1997; Efimov, 1993). A recent study has found that roughly 33% of all proteins have complete sequence coverage to a protein with known structure (Ekman et al., 2005). This kind of protein structure redundancy versus protein sequence variability is the cornerstone of homology modeling algorithms. It is quite likely that homology modeling will assume an increasingly important role in both biological and chemical applications with the advent of structural genomics initiatives around the world.
10.1.2History of Homology Modeling
Homology modeling techniques became important only after 1990 when hundreds of protein structures had already been deposited in the Protein Data Bank. Early modeling studies in the late 1960s and early 1970s frequently relied on the construction of hand-made wire and plastic models and only later depended on computer software (Cox and Bonanou, 1969; Tometsko, 1970). The first homology model was built simply by copying existing coordinates from a homologous protein and those nonidentical residues were then substituted by reassembling corresponding side chains (Browne et al., 1969). This approach, called rigid-body assembly, is still widely employed today with considerable success, especially when the proteins have sequence identity above 40% (Greer, 1980, 1981). The homology modeling method was pioneered in two studies by Browne et al. (1969) and Greer (1981). Browne et al. (1969) published the first homology model using an X-ray-derived structure as a template. They modeled bovine -lactalbumin on the three-dimensional structure of hen eggwhite lysozyme, where the pair sequence identity was about 39% and only deletions were considered as the polypeptide chain was shortened in -lactalbumin. Their prediction was proven generally correct later on when the structure of -lactalbumin was solved (Acharya et al., 1989).
McLachlan and Shotton (1971) modeled alpha-lytic proteinase of Myxobacter 495 based on the structures of both chymotrypsin and elastase, where the sequence identity between these two proteinases was only 18% and the alignment was fragmented by frequent gaps. When the structure of alpha-lytic proteinase was published (Brayer et al., 1979), it became clear that misalignment of the sequence with those of the known 3D structures led to incorrect regions, but portions of both domains were constructed correctly (Delbaere et al., 1979). This model demonstrated the difficulty in aligning sequences of limited similarity and in modeling variable, mainly loop regions.
Greer was the first to demonstrate the importance of modeling variable regions (1981). By abstracting approximate conformations from a family of homologous proteins of known structures, he could distinguish structurally conserved regions, which contain strong sequence homology, and structurally variable regions, which include all the insertions and deletions. By applying the structural distinction to new sequences, erroneous alignments of the sequences are greatly minimized. For each new aligned sequence, the structurally conserved regions can be constructed from
322 |
Zhexin Xiang |
any of the known structures. The construction of variable regions, however, is not straightforward. However, the conformations of loops of just oneor two-residue deletions or insertions can be extrapolated from one of the homologous structures. This approach was further applied to predict the structure of mammalian serine proteases based on a number of proteins from this family, including a variety of blood-serum, intestinal, and pancreatic proteins as well as a closely related bacterial enzyme (Greer, 1981).
Instead of deriving protein backbone structure from only one of its homologues, Taylor (1986) developed a method of generating templates for each part of protein to be modeled based on the conserved patterns observed in the known 3D structures of a family. The conserved templates were derived from a small number of related sequences of the known tertiary structures. The templates were then made more representative by aligning with other sequences of unknown structures. The specificity of the templates was demonstrated by their ability to identify the conserved features in known immunoglobulin and the related sequences but not in other sequences. However, assembling these conserved patterns into a complete structure requires the use of a force field and conformation sampling.
Due to the small number of protein structures available before the 1990s, the comparative modeling technique was not widely successful. The real development of homology modeling began in the mid-1990s with the progress of genome projects and the growth of the number of solved structures in the PDB. With the advent of structural genomics, the importance of homology modeling continues to grow. Although 25 years have passed since Greer’s pioneering work on comparative model building of mammalian serine protease in 1981, the basic technique used in today’s most advanced modeling programs remains almost the same, i.e., finding the closest homologues as the basis of modeling the query sequence. Recent efforts in comparative modeling have been concentrated on the discovery of distant homologues, the improvement of alignment accuracy, and especially the refinement of models by optimization of empirical energy functions.
10.1.3Accuracy and Applicability of Homology Modeling
Approximately 57% of all known sequences have at least one domain that is related to at least one protein of known structure (Pieper et al., 2002). The probability of finding a related known structure for a randomly selected sequence from a genome ranges from 30% to 65%, since a few genomes have received more research attention than others (Kelley et al., 2000; Teichmann et al., 1999; Fiser and Sali, 2003). The percentage is steadily increasing because more distinct folds are discovered each year, and because the number of different structural folds that proteins adopt is limited (Irving et al., 2001). Current estimates suggest that there are between 1000 and 5000 folds in the universe of compact globular proteins, with about 200 new folds realized annually from the structure deposition (Brenner et al., 1997). The number of known protein sequences is close to 2 million so far. Over 1.1 million proteins can readily have at least one of their domains reliably predicted with homology modeling
10. Homology-Based Modeling of Protein Structure |
323 |
methods. Given the rate of experimental structure determination, approximately 6000 proteins each year, it is arguable that homology modeling has already saved up to hundreds of years of human effort, though homology models often have low quality. In the next 10 years, structural genomics will possibly discover all protein distinct folds in Nature, making comparative modeling applicable to almost any protein sequence (Vitkup et al., 2001). The usefulness of comparative modeling is ever increasing as more proteins can be predicted with higher accuracy. The accuracy of homology modeling depends primarily on the sequence similarity between the target sequence and the template structure.
When the sequence identity is above 40%, the alignment is straightforward, there are not many gaps, and 90% of main-chain atoms can be modeled with an
˚
RMSD error of about 1 A (Sanchez and Sali, 1997). In this range of sequence identity, the structural difference between proteins mainly arises from loops and side chains. When the sequence identity is about 30–40%, obtaining correct alignment becomes difficult, where insertions and deletions are frequent. For sequence similarity in this range, 80% of main-chain backbone atoms can be predicted to RMSD
˚
3.5 A, while the rest of the residues are modeled with larger errors, especially in the insertion and deletion regions (Harrison et al., 1995; Mosimann et al., 1995; Yang and Honig, 2000; Sauder et al., 2000). Even in correctly aligned regions, loop modeling and side-chain placement pose difficulties (Bower et al., 1997; Rapp and Friesner, 1999). When the sequence similarity is below 30%, the main problem becomes the identification of the homologue structures, and alignment becomes much more difficult. For some sequences where the structures in the family are very conserved in evolution (e.g., kinase family), homology modeling can make predictions as accurate as low-resolution X-ray experiments even if the sequence identity is much less than 30% identity to the template (Yang and Honig, 1999; Petrey et al., 2003).
Even if homology modeling is generally much less accurate than experimental methods, it can still be helpful in proposing and testing hypotheses in molecular biology, such as predictions of ligand binding sites (Zhou and Johnson, 1999; Francoijs et al., 2000), substrate specificities (Jung et al., 2000; De Rienzo et al., 2000), function annotation, protein interaction pathways, and drug design (Nugiel et al., 1995; Sanchez and Sali, 1997). It can also provide starting models for solving structures from X-ray crystallography, NMR, and electron microscopy (Talukdar and Wilson, 1999; Ceulemans and Russell, 2004).
10.2 Procedures in Homology Modeling
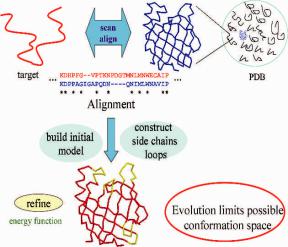
Given a protein sequence, successful homology modeling usually consists of the following steps as shown in Fig. 10.2: (1) identify the homologue of known structure from the Protein Data Bank; (2) align the query sequence to the template structure;
(3) build the model based on the alignment; (4) assess and refine the model. Each step may involve some errors.
324 |
Zhexin Xiang |
Fig. 10.2 Basic homology modeling protocol. Homology modeling starts by scanning the PDB for sequences similar to the target. The hit of highest sequence similarity is chosen as the template. Model for the target is then built based on the alignment between the target and template sequence, which will be subjected to further refinement.
10.2.1Homologue Detection and Alignment
Homology modeling starts from selection of homologues with known structures from the PDB. If the query sequence has high sequence identity (>30%) to the structure, the homology detection is quite straightforward which is usually done by comparing the query sequence with all the sequences of the structures in the PDB. This can often be achieved simply with the dynamic programming method (Needleman and Wunsch, 1970) and its derivatives (Smith and Waterman, 1981; Gotoh, 1982). The most popular software is BLAST (Altschul et al., 1997) ( http://www.ncbi.nlm.nih.gov/blast/) that searches sequence databases for optimal local alignments to the query. The BLAST program improves the overall speed of searches while retaining good sensitivity by breaking the query and database sequences into fragments, and initially seeking matches between fragments. The matched fragments are then extended in either direction in an attempt to generate an alignment with a score exceeding a particular threshold. The score based on substitution matrices reflects the degree of similarity between the query and the sequence being compared, capable of ranking the quality of each pairwise alignment. The BLAST program functions very well for alignment of sequences with high similarities. But when the sequence identity is well below 30%, homology hits from BLAST are not reliable. A number of alternative strategies have been developed. These include template consensus sequences (Taylor, 1986; Chappey et al., 1991) and profile analysis (Barton and Sternberg, 1990; Suyama et al., 1997; Lolkema and Slotboom, 1998). All these approaches, based
10. Homology-Based Modeling of Protein Structure |
325 |
on either multiple sequence or structure alignments, are more sensitive because the consensus sequences are more representative of the sequence family, and the profile reflects the conserved structural or functional preferences.
In the past several years, sequence profile methods have emerged as the primary approach in distant homology detection. Position-specific profile search methods such as PSI-BLAST (Altschul et al., 1997) and hidden Markov models (HMMs) (Krogh et al., 1994), as implemented in the SAM (Karplus et al., 1998) and HMMER (http://hmmer.wustl.edu) packages, have vastly improved the accuracy of sequence alignments and have extended the boundaries of detectable sequence similarity. Sequence profiles methods, e.g., PSI-BLAST, start from performing a pairwise search of the database. The significant alignments are then used by the program to construct a position-specific score matrix (PSSM). This matrix replaces the query sequence in the next round of database searching. The procedure may be iterated until no new significant alignments are found. The profile method can be further improved with information from multiple structure alignment, secondary structure prediction, and solvent accessibility. Since the structural information is more conserved than sequence, it may represent the crucial requirement, in the process of evolution, of residues at specific positions with respect to the stability and function of the structure as a whole. Although a major goal of this effort has been remote homologue detection, an important side benefit has been significant improvement in alignment quality, even at levels of sequence identity for which pairwise alignment methods are known not to work. This, in turn, has had a positive impact on the starting alignments used in homology modeling, and thus has the potential to extend the applicability of homology modeling to increasingly lower levels of sequence similarity. Indeed, perhaps the largest part of recent improvements in homology modeling can be traced directly to improvement in sequence alignment algorithms.
If multiple homologues from the PDB have been identified, the next step is to select one or a few templates that are most appropriate for building a model. Sequence similarity between the query and the template is usually considered as the primary criterion used to choose the best template. Higher sequence similarity often suggests a closer relationship in evolution, thus more conservation of structure, and vice versa. On the other hand, gaps (insertion or deletion) in the alignment have a severe impact on the quality of the model to be built, since gaps are regions where no templates are readily available to guide the model building process. Generally, insertions in the alignment are more difficult to handle than deletions, particularly for insertions of more than 10 residues, because modeling inserted residues is a mini ab initio problem. Thus, the second criterion is to choose an alignment that has fewer gaps and short insertions. Moreover, since sequences in the same family often share similar structure and function, templates that can be clustered into the same subfamily as the target are often favored. This can usually be achieved with construction of a multiple alignment and phylogenetic tree (Felsenstein, 1981; Retief, 2000). If the function of the protein to be modeled (target protein) is known, templates with similar functions should also be given more consideration. If possible, the
326 |
Zhexin Xiang |
environment (e.g., ligands, solvent, temperature, pH) at which the template structure was determined and the native environment of the target protein to be modeled should also be properly taken into account. A protein, such as calcium binding protein, can adopt quite different conformations in solvent under different environments (Mishig-Ochiriin et al., 2005). Thus, structures determined in environments similar to the physiological environment of the target are generally preferred. In addition, structures of higher quality are generally used, such as X-ray structures of high resolution and low R-factor, and NMR structures with sufficient constraints. In the end, structures that are most representative of the family should be used if all other criteria are identical. The trait can easily be calculated as the average RMSD of the structure to all other family members with known structures. The hypothesis is that the target protein is more likely to be similar to the most “typical” structure. Instead of relying on a single template, it can be advantageous to select multiple structures from one or several families. Multiple template structures may be aligned with different domains of the target, thus a composite model can be built with each domain based on the best template. It is also useful for modeling variable regions of a structure family, where the segment, which is not conserved, assumes multiple conformations, and the “best” model is assumed to have the lowest value of some empirical energy function. If the sequence identity is too low and there is no clear hit, a better approach is always to make multiple models with each model based on one template. Thus, the best model is determined by physical chemistryor statistics-based energy or a combination of both (Sippl, 1995; Petrey et al., 2003; also see Chapters 2 and 3).
In homology modeling, one of the most difficult and important tasks is to improve sequence–template alignment. Although profile methods have significantly improved alignment accuracy, manual inspection is often required to further improve the quality if the alignment is well below 30% identity with frequent gaps. This is because the current alignment software usually seeks an alignment of global optimality with an empirical scoring function that may misalign functionally important residues. Manual inspection of the alignment does not necessarily need to have the model actually built, since residue–residue interaction in the target sequence can easily be identified from their corresponding aligned positions in the template structure. There are several general rules to guide alignment tuning. First, charged residues in the target sequence should not be aligned with a buried residue in the template, unless it will form hydrogen bonds or salt bridges with another residue in the target; second, fragments of predicted secondary structures (alpha helix and beta sheet) in the target sequence should be aligned with the fragments of identical secondary structure characterization from the template; third, residues with known important functions, either for protein activity or structural stability, should be aligned with residues of similar functions in the template; fourth, insertions or deletions in the secondary structure regions should be pushed to the loop regions. Manual editing of the alignment is the most tedious part in homology modeling. A misalignment by
˚
only one residue position will result in an error of approximately 4 A in the model because the current homology-modeling algorithms generally cannot recover from errors in the alignment (Fiser and Sali, 2003).

10. Homology-Based Modeling of Protein Structure |
327 |
|
|
Table 10.1 Comparative modeling programs |
|
|
|
|
Programs |
Availability |
|
|
|
|
NEST |
http://trantor.bioc.columbia.edu/programs/jackal/ |
|
COMPOSER |
http://www-cryst.bioc.cam.ac.uk/ |
|
Tripos (COMPOSER) |
http://www.tripos.com/ |
|
CONGEN |
http://www.congenomics.com/congen/congen toc.html |
|
MODELLER |
http://guitar.rockefeller.edu/modeller/modeller.html |
|
InsightII (MODELLER) |
http://www.accelrys.com/ |
|
SWISS-MODEL |
http://www.expasy.ch/swissmod/SWISS-MODEL.html |
|
SCHRODINGER |
http://www.schrodinger.com |
|
WHATIF |
http://swift.cmbi.kun.nl/whatif/ |
|
SEGMOD |
http://www.bioinformatics.ucla.edu/genemine/ |
|
DRAGON |
rmunro@nimr.mrc.ac.uk |
|
ICM |
http://www.molsoft.com/ |
|
3D-JIGSAW |
http://www.bmm.icnet.uk/servers/3djigsaw/ |
|
Builder |
koehl@csb.stanford.edu |
|
PrISM |
http://trantor.bioc.columbia.edu/programs/PrISM/index.html |
|
|
|
|
10.2.2Model Building
Given the alignment between the query sequence and templates, there are generally four methods in model building depending on how the information in the known structures is transferred to the query sequence. In this section we are going to discuss the first three methods, i.e., rigid body assembly, segment matching, and spatial restraint, and leave our own approach (artificial evolution model building) to Section 10.3 for more detailed description. Table 10.1 shows the most widely used model building programs that are publicly available. Most of the programs were based on the rigid body assembly method, and some have been commercialized, e.g., COMPOSER (Sutcliffe et al., 1987a,b) in Tripos and MODELLER (Sali and Blundell, 1993) in InsightII. In addition to model building protocols, the programs also differ from each other in model refinement.
10.2.2.1 Model Building by Rigid Body Assembly
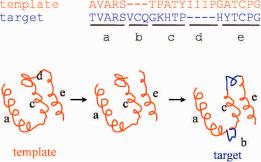
The simplest and most widely used method is called rigid body assembly (also called cut-and-paste method) as shown in Fig. 10.3. This method was initiated by Greer in 1981 and is still widely used, e.g., in the software packages PriSM (Yang and Honig, 1999), Congen (Bruccoleri, 1993), and COMPOSER (Sutcliffe et al., 1987a,b). It starts from identification of the conserved and variable regions of the templates. The identification can often be achieved from the superimposed template structures. Conserved regions are evident from the multiple structure alignment, that is, the RMSDs (root mean square distance) among the fragments are relatively small, and variable regions are usually in loops with frequent gaps in the structural alignment. A
328 |
Zhexin Xiang |
Fig. 10.3 Rigid body assembly with single template. Red and blue denote template and model structure, respectively. Conformations of aligned segments a, c, and e are directly transferred to the model; segment b is a new conformation inserted between a and c, which is obtained from either ab initio sampling or database searching; segments c and e are fused following the deletion of segment d.
framework for the superimposed templates can be calculated by averaging the atom coordinates of the structurally conserved regions. The averaging is often weighted based on the sequence similarity of the target sequence to the templates, higher sequence similarity carrying larger weight. The core residues of the target model, i.e., residues aligned with conserved regions of the templates, obtain their mainchain coordinates from the closest conserved segment (in terms of RMSD to the framework) from the template, or from the segment whose template has highest sequence identity to the target. The model is constructed by fitting the core rigid bodies onto the framework. The unconserved, or loop, region is then constructed either with ab initio approach, or by searching a database for structures that fit the anchor core regions and have a compatible sequence (Topham et al., 1993). The side chains are modeled based on their intrinsic conformational preferences and on the conformations of the equivalent side chains in the template structures (Sutcliffe et al., 1987a,b). If a single template is chosen, the model construction is straightforward, copying coordinates of aligned residues from the template to the model, and connecting broken segments with database searching or ab initio sampling as mentioned earlier. For strong sequence homologues, a single template is sufficient; but for weakly homologous templates, a framework based on weighted averaging over multiple templates is often more reliable.
10.2.2.2 Model Building by Segment Matching
Segment matching (Levitt, 1992), which has been adopted in SegMod software, is based on the finding that most hexapeptide segments of protein structure can be clustered into only 100 structurally different classes (Unger et al., 1989). Homology model construction relies on approximate positions of conserved atoms from the templates as “guiding positions” to calculate the coordinates of other atoms. The guiding positions usually correspond to the atoms of the segments that are conserved