

Computational Methods for Protein Structure Prediction & Modeling V1 - Xu Xu and Liang
.pdf298 |
Jun-tao Guo et al. |
intrinsic property of proteins under slightly denatured concentrated conditions stemming from the interplay between protein geometry, backbone hydrogen bonding, and hydrophobicity (Dobson, 1999, 2003). The weak dependence of protofilament structure on sequence and the great speed of low-resolution model simulations make this approach well suited to examinations of aggregation kinetics on a molecular level.
Low-resolution model studies of protein aggregation can be differentiated based on several characteristics: on-lattice versus off-lattice, two-dimensional versus threedimensional, few chains versus many chains, short chains versus long chains, simple interaction potential versus complex interaction potential, amorphous aggregate versus ordered (fibrillar) aggregate, and whether or not the structure of the monomer in the fibrillar state differs or is the same as the structure of the monomer in the isolated native state. As will be seen from the discussion below, the models have gotten more complex and more realistic as time has progressed.
Low-resolution lattice models: The earliest low-resolution model of protein aggregation was a lattice model introduced in 1994 by Patro and Przybycien (Patro and Przybycien, 1994; Patro et al., 1996). They modeled a system of proteins as a collection of hexagons with polar and nonpolar surface sites moving on a two-dimensional lattice. Their aim was to learn how surface characteristics influence the formation kinetics and structure of the observed aggregates. Since their monomers were essentially in the folded state, they could not explore how folding and unfolding impacts aggregation. This early work spurred other groups to apply lattice models in the study of protein aggregation, but with the caveat that the model proteins were allowed to fold and unfold.
The lattice models can be distinguished according to the size of the “alphabet” used to represent the various residues. The simplest of the lattice models is the two- letter-alphabet “HP model” introduced by Lau and Dill (1989), in which a protein is modeled as a chain of hydrophobic (H) and polar (P) residues arranged in a specific sequence. Nonbonded H beads attract each other with strength ε to account for the hydrophobic effect while nonbonded P–P and H–P interactions are set equal to zero. This mimics the tendency of hydrophobic residues to bury themselves in the protein interior in order to avoid contact with water. Even this simple “two-letter- alphabet” model allows investigators to extract (via Monte Carlo simulation or exact enumeration) the general theoretical principles that underlie the connection between a protein’s sequence and its native structure, folding pathways, kinetics, and the like.
A common theme running through many of the lattice simulations is the nature of the monomer structure within the ordered aggregate. The question is whether the protein remains soluble in its native state conformation under all conditions, adopts its native state structure within the ordered aggregate, or adopts an alternate structure (the so-called prionlike structure). This theme was explored by Giugliarelli et al. (2000) using a two-dimensional HP model; they found that the answer to the previous question depends sensitively on the amino acid composition. In fact, the most stable proteins were those whose fraction of hydrophobic residues is similar to that found in naturally occurring proteins. Harrison et al. studied the formation of
9. Modeling Protein Aggregate Assembly and Structure |
299 |
dimers using twoand three-dimensional HP lattice models (Harrison et al., 1999) and a simple two-dimensional four-letter-alphabet lattice model (Harrison et al., 2001)with residues of types H (hydrophobic), P (polar), A, and B, where A and B have a particular affinity for each other. They observed that the protein sequences that were marginally stable as monomers were more likely to be stabilized in an alternate conformation by the multimeric interactions in a dimer aggregate, which was at an energy minimum. The dimers that were rich in -sheet structure were more likely to propagate their conformations onto other chains, hence the term “prionlike.”
The HP model has been used by a number of groups to learn how the structure of the ordered aggregate depends on the protein sequence, concentration, and temperature. Istrail et al. (1999) studied the dependence of the aggregation of two model proteins on the hydrophobic/hydrophilic sequence and composition along the chain as well as chain packing fraction (essentially the concentration). Not surprisingly, the higher the number of hydrophilic residues, the lower the aggregation propensity is. Dima and Thirumalai (2002) used a three-dimensional HP lattice model containing two proteins to probe how the conformational change from a compact monomeric state to an oligomeric -sheet state depends on temperature and concentration. They observed three distinct ordered states, only one of which contained the native state.
Other investigators have adopted much larger alphabets to describe their lattice proteins. The 20-letter alphabet proposed by Miyazawa and Jernigan (1985) is quite popular. In this model the interresidue interaction potentials are estimated from the numbers of interresidue contacts observed in crystal structures in the PDB. Broglia et al. (1998), Bratko and Blanch (2001), Cellmer et al. (2005), and Leonhard et al. (2003) have all used this alphabet in their Monte Carlo simulation explorations of how protein sequence and concentration influence the aggregation kinetics and thermodynamics of systems containing a small number of chains.
All of the work mentioned above was limited to only a few chains which is not enough to fully explore the competition between protein folding and aggregation. In contrast, the simulations of Combe and Frenkel (2003), Toma and Toma (2000), and Hall and co-workers (Gupta and Hall, 1997; Gupta et al., 1998; Nguyen and Hall, 2002) were truly multichain systems. Combe and Frenkel performed Monte Carlo simulations on a system containing twenty 8-mer peptides whose interactions were modeled using the “Go model,” a model in which the interactions are chosen to favor the known native state. Even though Go models introduce a strong bias toward the isolated chain’s native state, they can still be used profitably to explore the kinetics of protein aggregation and the competition between folding and aggregation. Toma and Toma conducted lattice Monte Carlo simulations on systems containing as many as twenty 12-mer HP peptides of three different sequences in an effort to learn how the sequence and concentration affect the formation of an ordered state.
Hall and co-workers (Gupta and Hall, 1997; Gupta et al., 1998; Nguyen and Hall, 2002) conducted simulations on systems containing as many as 40 twodimensional 16-mer HP chains of a single sequence in an effort to learn how the protein concentration, denaturant concentration, and temperature affected the refolding yield and the kinetics of the aggregation pathway. Denaturant concentration was
300 |
Jun-tao Guo et al. |
modeled implicitly; the stronger the interaction between the hydrophobic residues, the weaker the denaturant concentration. Since their aim was to learn how to optimize protein folding yield during recovery from inclusion bodies, they focused primarily on aggregation into nonstructured states. Nguyen and Hall (2002) performed simulations that mimicked four methods of thermal protein renaturation used in the lab: dialysis, dilution (or diafiltration), quenching, and pulse renaturation (fed-batch operation). Based on the simulation results, a strategy for rapidly obtaining high refolding yields was suggested which involved instantaneous removal to intermediate denaturant concentrations followed by dialysis to the final state.
Low-resolution models—off lattice: Off–lattice low-resolution protein models have been used extensively in the past decade to simulate the folding of an isolated protein. They are just beginning to be used in studies of aggregation. In the simplest of these models, a protein is represented as a flexible chain of spheres (think pearl necklace), with each sphere representing a single residue. The interactions between the spheres are represented by energy functions that can be divided roughly into three categories: (1) Go-type potentials in which the parameters are chosen to favor the protein’s known native state (Go and Taketomi, 1978; Taketomi et al., 1975),
(2) potentials based on the relative hydrophobicity of the side chains as measured by various hydrophobicity scales (and sometimes on the partial charge), and (3) knowledge-based potentials like the Miyazawa–Jernigan potential (Miyazawa and Jernigan, 1985) in which statistical data on residue–residue contacts from the PDB are used to infer side chain/side chain potentials. Local interaction potentials are also used to maintain steric and angular constraints.
Jang et al. (2004a,b) used an off-lattice low-resolution protein model to investigate the thermodynamics and kinetics associated with the folding, aggregation, and fibrillization of -strand peptides. The goal was to learn why multiprotein systems sometimes form ordered aggregates and sometimes form amorphous aggregates. Their off-lattice protein model consisted of 39 single-sphere residues interacting intramolecularly via a square–well Go potential (Go and Taketomi, 1978; Taketomi et al., 1975) that favored the four-strand antiparallel -sheet native state and intermolecularly via a second square–well Go potential that favored the formation of a tetrameric -sheet complex (a model fibril). In fact, the intraand intermolecular interactions could be interpreted as mimicking hydrogen bonding and hydrophobic interactions, respectively. The ratio of the strengths of the intraand intermolecular potentials was varied. Discontinuous MD simulations (described in a later section) were performed on systems containing a single protein and four proteins to see how the equilibrium properties, folding pathways, and kinetics varied as a function of the ratio of the intraand intermolecular interactions and the temperature. A phase diagram was constructed showing which monomer states and which tetrameric complex states were stable at various temperatures and interaction strength ratios. At high temperatures the four-peptide system formed monomers but at low temperatures the system assembled into tetrameric -sheet complexes that were either partially ordered , ordered, or highly ordered, depending on the relative strength of
9. Modeling Protein Aggregate Assembly and Structure |
301 |
the interand intramolecular interactions. The most ordered structures were found at intermediate values of this ratio. This implies that for ordered aggregates to form, there must be a balance between the hydrogen bonding interactions that hold the-strands together in a -sheet and the hydrophobic interactions that hold the sheets together. If either is too large, disordered rather than ordered aggregates will form at high concentrations.
Ding et al. (2002a,b) also used an off-lattice low-resolution model to study the aggregation of a system of eight model Src SH3 domain proteins. Each amino acid residue along the flexible backbone chain was represented by a single backbone sphere and a single side-chain sphere interacting via a Go potential designed to recover the protein’s native state. A fibrillar double -sheet structure was observed with inter- -strand spacing and inter- -sheet spacing similar to those observed in experiments.
Intermediate-resolution protein models: In recent years a new class of protein folding models has been introduced, called intermediate-resolution folding models (Derreumaux, 1999; Liwo et al., 1997; Takada et al., 1999; Wallqvist and Ullner, 1994). The idea here was to add more realistic features to the low-resolution protein folding models in the hopes that this would allow a priori prediction of the native state structure of specific proteins based solely on their amino acid sequence. The number of spheres used to represent protein geometry was increased from one to as many as seven. The energy functions were expanded to include not only the three categories described for the low-resolution off-lattice models but also hydrogen-bonding potentials, multibody terms, burial terms (in which the strength of the hydrophobic interaction depends on the extent of burial), and special potentials for disulfide bonds and proline.
Intermediate-resolution protein models are now being used to study aggregation and fibril formation. In 2001, Hall and co-workers (Smith and Hall, 2001a,b,c) introduced an implicit-solvent intermediate-resolution protein model, which they subsequently named PRIME (Nguyen et al., 2004). This model is simple enough to allow the simulation of systems containing many proteins over long time scales, yet contains sufficient molecular detail to mimic real protein dynamics. The level of molecular detail in the protein representation and interaction potential is reduced just to the point at which the key physical features governing protein fibrillization remain and the other features are neglected. In PRIME, each amino acid is composed of four spheres: a three-sphere backbone comprised of united atom NH, C , and C=O and a single-sphere side chain (CH3 for alanine), all with realistic diameters and bond lengths. Ideal backbone bond lengths, bond angles, C –C distances, and residue L- isomerization are maintained by imposing a series of pseudo bonds whose lengths fluctuate within a tolerance of 2% about the specified values.
All forces in PRIME are modeled by either hard-sphere or square-well potentials with realistic diameters. This was done so as to allow the use of discontinuous MD (DMD) simulation, a very fast alternative to traditional MD simulation that is applicable to systems of molecules interacting via discontinuous potentials, e.g.,
302 |
Jun-tao Guo et al. |
hard-sphere and square-well potentials (Rapaport, 1978, 1979). Instead of solving Newton’s equation of motion at regular spaced time intervals, as in traditional MD, DMD is event-driven. Since discontinuous potentials exert forces only when particles collide (unlike continuous potentials such as the Lennard–Jones potential), the position and velocity of each molecule after a collision can be determined exactly, as opposed to numerically. This imparts great speed to the algorithm, allowing sampling of much wider regions of conformational space, longer time scales, and larger systems than in traditional MD.
The solvent in PRIME is modeled implicitly by factoring its effect into the energy function as a potential of mean force. Interactions between hydrophobic side chains are represented by a square-well potential; interactions between polar side chains or between polar and hydrophobic side chains are represented by a hard sphere interaction. Hydrogen bonding between amide hydrogen atoms and carbonyl oxygen atoms is represented by a directionally dependent square-well attraction of strength between NH and C=O united atoms. (The angle between the “virtual” N–H and C=O vectors can be determined from knowledge of the locations of the adjacent united atoms along the chain.) The strength of the hydrophobic interaction is fixed at a fraction of the strength of the hydrogen bonding interaction; this fraction is the only adjustable parameter in the model.
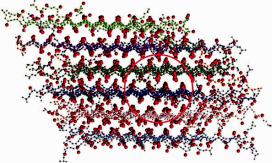
By combining PRIME with DMD, Nguyen and Hall (2004a, 2005) were able to simulate the spontaneous formation of ordered aggregates, essentially protofilaments, in model systems containing 48 to 96 polyalanine (Ac-KA14K-NH2) peptides (Fig. 9.3). Polyalanine was chosen for study because synthetic polyalanine-based peptides, which form -helical structures at low temperatures and low peptide concentrations, had been found experimentally to form -sheet complexes (fibrils) in vitro at high temperatures and high peptide concentrations. All simulations started from a random coil configuration equilibrated at a high temperature and then slowly cooled to the temperature of interest so as to minimize kinetic trapping. These simulations took approximately 40 hours on an AMD Athlon MP 2200+ single-processor workstation. The simulation results showed that the populations of -helices, amorphous aggregates, -sheets, and fibrils were highly dependent
Fig. 9.3 A closeup snapshot of the 96-peptide fibrillar structure of polyalanine.
9. Modeling Protein Aggregate Assembly and Structure |
303 |
on temperature and peptide concentration, in qualitative agreement with the experimental results of Blondelle and co-workers (Blondelle et al., 1997; Perez-Paya et al., 1996) on Ac-KA14K-NH2 peptides. The fibrils observed in the simulations mimicked the structural characteristics observed in experiments in that most of the fibrillar peptides were arranged in an in-register parallel orientation, with intraand intersheet distances similar to those observed in experiments. The simulations revealed that Ac-KA14K-NH2 fibril formation is nucleation dependent, which is similarly observed in experimental studies. The formation of small fibrils was preceded by the appearance of small amorphous aggregates, then -sheets, and finally rapid growth of a stable fibril. A phase diagram in the temperature–concentration plane was mapped out delineating the regions where random coils, -helices, -sheets, fibrils, and amorphous aggregates are stable (Nguyen and Hall, 2004b).
Models similar to PRIME have been developed by Stanley, Ding, Dokholyan, Teplow (Ding et al., 2003; Urbanc et al., 2004a,b) and co-workers for use with DMD (which they call “discrete molecular dynamics” as opposed to “discontinuous molecular dynamics”). Their main focus has been on the aggregation of amyloid, A , the protein whose oligomerization and fibrillization have been linked to Alzheimer’s disease. The A peptide is represented with a four bead per residue model as in PRIME. Their side chain representation and energy function are more complex than used in PRIME for polyalanine in order to account for the differences between all 20 amino acid residues. There are four types of side chains: neutral, charged hydrophilic, hydrophilic, and hydrophobic. The hydropathy of the various side chains is assigned according to the scale of Kyte and Doolittle (1982). In addition to having an attraction between hydrophobic side chains, they include a repulsive interaction between uncharged hydrophilic side chains and either charged or uncharged side chains. The side chain for glycine residues is absent.
This model is being used to tackle the difficult question of why A (1–42) is so much more amyloidogenic than A (1–40). A (1–42) is more likely to be associated with the early onset forms of Alzheimer’s, with increased risk for getting Alzheimer’s disease, with enhanced neurotoxicity, and with faster formation of fibrils in vitro. Urbanc et al. (2004a,b) conducted DMD simulations of systems containing 32 A (1–40) and 32 A (1–42) peptides starting from a system of random coil A monomers. Although they did not observe ordered structures in their simulations, they did observe important early events in the aggregation process including monomer folding and assembly into disordered oligomers of various sizes. Analysis of their results indicates that there are significant differences between the oligomer size distributions of A (1–40) and A (1–42), with A (1–40) more likely to form dimers and A (1–42) more likely to form pentamers, in agreement with in vitro size distribution studies. The A (1–42) peptide was likely to form a turn at Gly37–Gly38, whereas the A (1–40) was not. The structural differences between the conformations of the A (1–40) and A (1–42) oligomers suggest that the hydrophobic core of the A (1–42) pentamer is more exposed than that of the A (1–40) pentamer, and is therefore likely to form larger oligomers. This may have implications for the biology of Alzheimer’s disease since some believe that it is at the oligomer/protofibril level
304 |
Jun-tao Guo et al. |
that A is most toxic with A (1–42) being more toxic than A (1–40) (Caughey and Lansbury, 2003).
9.5 Summary
In summary, high-resolution structural characterization of protein aggregates using classical approaches, such as X-ray crystallography or solution NMR, has been hampered due to the insolubility and noncrystalline nature of the aggregates. Encouraging results have begun to emerge, however. Two high-resolution, detailed structures, both involving crystal packing with “infinite -sheet” (Schiffer et al., 1985) characteristics, have been obtained from crystals of amyloidogenic peptides (Makin et al., 2005; Nelson et al., 2005). One peptide is a seven-residue fragment of a yeast prion known as Sup35 (Nelson et al., 2005). The other one is a designed 12mer peptide containing two KFFE motifs separated by an AAAK motif (Makin et al., 2005). Experimental approaches as discussed in Section 9.3, including fiber diffraction, electron microscopy, hydrogen–deuterium exchange, solid-state NMR, limited proteolysis, electron paramagnetic resonance spectroscopy, and various chemical approaches, have yielded valuable information about the possible conformations of aggregate structure. But these low-resolution data are not sufficient to establish a high-resolution structure of protein aggregates, without which it will be difficult to address some of the fundamental questions regarding the molecular mechanism of aggregate assembly and the detailed interand intramolecular interactions that stabilize protein aggregates. Computational approaches can use the low-resolution experimental data to advance our understanding of the structure of protein aggregates and amyloid fibrils. All of the models constructed so far using computational approaches are motivated by or rely on experimental observations, and are validated using molecular dynamic simulations, a common technique for testing the stabilities of the models.
Considering that the insoluble nature of amyloid fibrils makes it hard to obtain high-resolution, detailed structural information, computational approaches should play a significant role in our efforts to solve the aggregate structure. Although computational methods are making strides in deciphering the mechanism of fibrillogenesis and the fibril core structure, many challenging issues need to be addressed in the future. First, computational studies of the aggregation process currently only apply lowto intermediate-resolution models. Novel ideas are clearly needed to investigate the process at a higher level given current computation capability. Second, it is well known that all amyloid fibrils share the common crossstructure, but the structural details might vary from sequence to sequence. Both antiparallel and parallel -sheet organizations have been suggested for the core structure from solid-state NMR studies. Moreover, recent studies have revealed that different growth conditions applied to the same peptide molecule can yield fibrils with distinct morphologies and possibly with different neurotoxicities (Petkova et al., 2005). Most importantly, amyloid fibril morphology correlates with internal architecture of the fibril, such
9. Modeling Protein Aggregate Assembly and Structure |
305 |
as side-chain packing arrangements, and the sequences involved in the -structure. Therefore, it is highly likely that there exist variations of amyloid folding motifs. Computational methods should be able to simulate the various structures under different conditions. In addition, comparative studies of amyloid fibril models formed by different amyloid proteins should be done in the future to elucidate the general principles regarding how specific interactions stabilize the fibril structures. Probably the most challenging issue in amyloid fibril structure modeling is the modeling of the packing patterns and detailed interactions among protofilaments, which might vary from fibril to fibril (Jimenez et al., 2002). Last but not least, we should also improve structural modeling techniques in such a way that new experimental data can be incorporated into the model as constraints. With the advance in computation speed and capability and the help from experimental observations, in the near future, we should be able to combine modeling studies with peptide assembling simulations for a better understanding of the process of amyloid fibril formation and detailed structure of the fibrils.
Suggested Further Reading
A special issue of the review journal Accounts of Chemical Research on amyloid (Vol 89, Issue 9, Sept., 2006) contains a number of articles on aspects of amyloid structure. Recent methods for analysis of amyloid structure, as well as some computational methods, are described in detail in Methods in Enzymology, Amyloid, Prions, and Other Protein Aggregates (R. Wetzel and I. Kheterpal, Eds.), Academic Press, San Diego, 2006, Volumes 412 and 413. The review by Zanuy et al. (2004) provides insights on amyloid structural formation and assembly through computational approaches.
Acknowledgments
The authors acknowledge support from the National Institutes of Health (R01 AG18927 to R.W., Y.X., and J.T.G.; AG18416 to R.W.; and R01 GM056766 to C.K.H.), National Science Foundation (DBI-0354771 to Y.X. and J.T.G.), and the Georgia Cancer Coalition (to Y.X. and J.T.G.).
References
Anfinsen, C.B. 1973. Principles that govern the folding of protein chains. Science 181:223–230.
Baker, D., Sohl, J.L., and Agard, D.A. 1992. A protein-folding reaction under kinetic control. Nature 356:263–265.
Balbirnie, M., Grothe, R., and Eisenberg, D.S. 2001. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA 98:2375–2380.
306 |
Jun-tao Guo et al. |
Benyamini, H., Gunasekaran, K., Wolfson, H., and Nussinov, R. 2003. Beta2microglobulin amyloidosis: Insights from conservation analysis and fibril modelling by protein docking techniques. J. Mol. Biol. 330:159–174.
Benzinger, T.L., Gregory, D.M., Burkoth, T.S., Miller-Auer, H., Lynn, D.G., Botto, R.E., and Meredith, S.C. 1998. Propagating structure of Alzheimer’s beta- amyloid(10-35) is parallel beta-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA 95:13407–13412.
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242.
Bhattacharyya, A.M., Thakur, A., and Wetzel, R. 2005. Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction.
Proc. Natl. Acad. Sci USA 102:15400–15405.
Bitan, G., and Teplow, D.B. 2004. Rapid photochemical cross-linking—A new tool for studies of metastable, amyloidogenic protein assemblies. Acc. Chem. Res. 37:357–364.
Blake, C.C., Geisow, M.J., Oatley, S.J., Rerat, B., and Rerat, C. 1978. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by
˚
Fourier refinement at 1.8 A. J. Mol. Biol. 121:339–356.
Blondelle, S.E., Forood, B., Houghten, R.A., and Perez-Paya, E. 1997. Polyalaninebased peptides as models for self-associated beta-pleated-sheet complexes. Biochemistry 36:8393–8400.
Bratko, D., and Blanch, H.W. 2001. Competition between protein folding and aggregation: A three-dimensional lattice-model simulation. J. Chem. Phys. 114:561– 569.
Broglia, R.A., Tiana, G., Pasquali, S., Roman, H.E., and Vigezzi, E. 1998. Folding and aggregation of designed proteins. Proc. Nat. Acad. Sci. USA 95:12930– 12933.
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C.M., and Stefani, M. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416:507–511.
Caughey, B., and Lansbury, P.T. 2003. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26:267–298.
Cellmer, T., Bratko, D., Prausnitz, J.M., and Blanch, H. 2005. Thermodynamics of folding and association of lattice-model proteins. J. Chem. Phys. 122:174908.
Chan, H.S., and Dill, K.A. 1990. Origins of structure in globular-proteins. Proc. Nat. Acad. Sci. USA 87:6388–6392.
Chan, W., Helms, L.R., Brooks, I., Lee, G., Ngola, S., McNulty, D., Maleeff, B., Hensley, P., and Wetzel, R. 1996. Mutational effects on inclusion body formation in the periplasmic expression of the immunoglobulin VL domain REI. Fold. Des. 1:77–89.
9. Modeling Protein Aggregate Assembly and Structure |
307 |
Chaney, M.O., Webster, S.D., Kuo, Y.M., and Roher, A.E. 1998. Molecular modeling of the Abeta1-42 peptide from Alzheimer’s disease. Protein Eng. 11:761–767.
Chen. S., Ferrone, F., and Wetzel, R. 2002. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA 99:11884– 11889.
Chick, H., and Martin, C.J. 1910. On the “heat coagulation” of proteins. J. Physiol. 40:404–430.
Chowdhry, V., and Westheimer, F.H. 1979. Photoaffinity labeling of biological systems. Annu. Rev. Biochem. 48:293–325.
Cleland, J.L., Powell, M.F., and Shire, S.J. 1993. The development of stable protein formulations: A close look at protein aggregation, deamidation, and oxidation.
Crit. Rev. Ther. Drug Carrier Syst. 10:307–377.
Coles, M., Bicknell, W., Watson, A.A., Fairlie, D.P., and Craik, D.J. 1998. Solution structure of amyloid beta-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 37:11064– 11077.
Collins, S.R., Douglass, A., Vale, R.D., and Weissman, J.S. 2004. Mechanism of prion propagation: Efficient amyloid growth in the absence of oligomeric intermediates. PLoS 2:1582–1590.
Colon, W., and Kelly, J.W. 1992. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31:8654–8660.
Combe, N., and Frenkel, D. 2003. Phase behavior of a lattice protein model. J. Chem. Phys. 118:9015–9022.
Creighton, T.E. 1992. Protein folding. Up the kinetic pathway [news; comment]. Nature 356:194–195.
De Bernardez Clark, E., Schwarz, E., and Rudolph, R. 1999. Inhibition of aggregation side reactions during in vitro protein folding. Methods Enzymol 309:217–236.
Del Mar, C., Greenbaum, E., Mayne, L., Englander, S.W., and Woods, V.L., Jr. 2005. Amyloid structure: alpha-synuclein studied by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA 102: 15477–15482.
Derreumaux, P. 1999. From polypeptide sequences to structures using Monte Carlo simulations and an optimized potential. J. Chem. Phys. 111:2301–2310.
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P., and Aronin, N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993.
Dill, K.A. 1990. Dominant forces in protein folding. Biochemistry 29:7133–7155. Dill, K.A., and Chan, H.S. 1997. From Levinthal to pathways to funnels. Nat. Struct.
Biol. 4:10–19.
Dima, R.I., and Thirumalai, D. 2002. Exploring protein aggregation and selfpropagation using lattice models: Phase diagram and kinetics. Protein Sci. 11:1036–1049.
Ding, F., Borreguero, J.M., Buldyrey, S.V., Stanley, H.E., and Dokholyan, N.V. 2003. Mechanism for the alpha-helix to beta-hairpin transition. Proteins Struct. Funct. Genet. 53:220–228.