

Глава V Физико-химические методы анализа
Основные направления развития аналитческих методов. Одним из направлений развития физико-химических методов анализ является их компьютеризация. В настоящее время практически все приборы, используемые в аналитических целях, оснащены компьютерами, это значительно сокращает время анализа, повышает его надёжность и информативность. Например, если без компьютера полная обработка УФ спектра требовала несколько часов, с компьютером подобные операции требуют несколько минут и даже меньше. Поскольку современные компьютеры допускают использование осень больших баз данных, то аналоговое отнесение полос в спектрах представляет собой довольно простую задачу. Без компьютера это также требовало значительного времени.
Другое направление усовершенствования физико-химических методов анализа – это совмещение различных методов на базе одного прибора. В частности, уже довольно давно серийно выпускаются хроматомасс- спектрометры. Эти приборы совмещают масс-спектроскопию и хроматографию.
Кроме того, приборы совершенствуются и в чисто техническом аспекте, например, качество ЯМР-спектров напрямую зависит от частоты магнитного поля в рабочем магните. Если в первых ЯМР-спектрометрах частота достигала порядка 100 мГц, то в современных спектрометрах она составляет уже тысячи мГц.
Оптические методы анализа. Оптические методы анализа для качественного и количественного определения веществ используют их взаимодействие со светом. Известно, что свет это электромагнитные колебания разной частоты, чем больше частота электромагнитного излучения, тем больше его энергия. Для аналитических целей используется практически весь спектр электромагнитного излучения: от излучений очень высоких энергий
(γ-излучение) в рентгеноструктурном анализе (РСА) до радиоволн метрового диапазона в ЯМР- и ЭПР-спектрах.
Наиболее распространены (и наиболее дёшевы) в аналитической практике ИК-, УФ спектроскопия и спектроскопия в видимой области (фотокалометрия). Электромагнитные волны, как и любой аналитический сигнал характеризуется качественным (частота) и количественным (интенсивность) параметрами.
Электронные абсорбционные спектры поглощения. Электронные абсорбционные спектры поглощения или ультрафиолетовые спектры (УФ спектры) исторически являются одним из самых давних и самых распространённых физико-химических методов исследования и определения химических соединений. Приставка «ультра»-, т. е. сверх- означает, что используется излучение, которое по энергии превышает видимый фиолетовый свет:
|
← Красн.,Оран.,Желт.,Зелён.,Голуб., Син., Фиол. → |
На схеме между стрелками представлено излучение, которое определяет видимый свет, энергия излучения увеличивается слева направо. Излучение правее фиолетового цвета называется ультрафиолетовым излучением, источники такого излучения и применяются в УФ спектрометрах, излучение левее видимого красного цвета (по энергии меньшее) называется инфракрасным излучением, источники такого излучения используются в инфракрасных спектрометрах (ИК спектрометрах). Видимая область также используется в анализе и называется фотометрия и применяется для исследования и определения окрашенных соединений.
Основной закон светопоглощения. Атом, ион, радикал, молекула, поглощая квант света, переходит в более высокое (возбуждённое) энергетическое состояние. Обычно это бывает переход с основного, невозбуждённого уровня на первый возбуждённый уровень. Вследствие поглощения излучения при прохождении его через слой вещества интенсивность излучения уменьшается и тем больше, чем больше концентрация вещества.
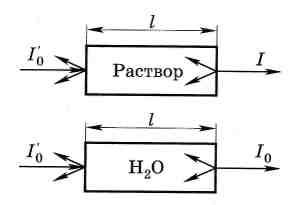
Закон Бугера – Ламберта – Бера (основной закон светопоглощения) связывает уменьшение интенсивности света, прошедшего через слой светопоглощающего вещества, с концентрацией вещества и толщиной поглощающего слоя. Чтобы учесть потери света на отражение и рассеяние, сравнивают интенсивности света, прошедшего через исследуемый раствор и растворитель (кювету сравнения, рис. 5.1). При одинаковой толщине слоя в кюветах
из одинакового материала, содержащих один и тот же растворитель, потери на отражение света будут примерно одинаковы у обоих пучков и уменьшение интенсивности света будет зависеть от концентрации вещества.
Уменьшение интенсивности света, прошедшего через раствор, характеризуется коэффициентом пропускания (или просто пропусканием) Т:
|
Т = I /I0, |
где I и I0 - соответственно интенсивности света, прошедшего через раствор и растворитель.
Взятый с обратным знаком логарифм Т называется оптической плотнностью А:
|
|
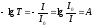 .
.
|
|
|
Рис. 5.1. Прохождение света через окрашенный раствор и растворитель |
Уменьшение интенсивности света при прохождении его через раствор подчиняется закону Бугера – Ламберта – Бера (см. выше):
|
I = I0 . 10-εlc, |
или
|
I / I0 = 10-εlc, |
или
|
-lgТ = А = -εlc, |
(5.1) |
где ε – молярный коэффициент поглощения; l – толщина светопоглощающего слоя; с – концентрация раствора.
Физический смысл ε становится понятным, если принять l = 1см и с = 1 моль/л, тогда А = ε. Следовательно, молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине слоя 1 см.
Ограничения и условия применимости закона Бугера – Ламберта - Бера. В соответствии с уравнением (5.1) зависимость оптической плотности от концентрации графически выражается прямой, выходящей из начала координат. Опыт показывает, однако, что линейная зависимость соблюдается не всегда. При практическом применении закона Бугера – Ламберта – Бера необходимо учитывать следующие ограничения:
1) Закон справедлив для монохроматческого света, т. е. для какой-либо одной (отсюда моно) длины волны. Чтобы учесть это ограничение, в уравнение (5.1) вводят индекс λ, которым в электронной спектроскопии обозначается длина волны, при которой происходит поглощение света:
|
Аλ = ελlc. |
(5.2) |
Здесь индекс λуказывает, что величиныAиεотносятся к монохроматическому излучению с длиной волныλ.
2) Коэффициент ε в уравнении (5.1) зависит от показателя преломления среды.
Более точное уравнение закона Бугера – Ламберта – Бера имеет вид:
|
|
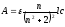 ,
,где n– показатель преломления.
Если концентрация раствора сравнительно невелика, его показатель преломления остаётся таким же, каким он был у чистого растворителя, и отклонений от закона по этой причине не наблюдается.
Изменение показателя преломления в высококонцентрированных растворах может явиться причиной отклонений от основного закона светопоглощения.
3) Температура при измерениях должна оставаться постоянной.
4) Пучок света должен быть параллельным.
5) Уравнение (5.1) соблюдается только для систем, в которых светопоглощающими центрами являются частицы лишь одного сорта. Если при изменении концентрации будет изменятся природа частиц, например, в результате кислотно-основного взаимодействия, то зависимость А от с не будет, в общем случае, линейной, т. к. молярные коэффициенты у реагентов и продуктов реакции, как правило, разные.
6) Интенсивность рассеянного света, возникающего в оптической системе прибора, должна быть сведена к минимуму.
Вид спектров поглощения. Свет поглощается раствором избирательно: при некоторых длинах волн светопоглощение происходит интенсивно, а при некоторых свет не поглощается. Интенсивно поглощаются кванты света, энергия которых hν точно равна разнице энергий между соответствующими энергетическими уровнями и вероятность такого возбуждения отлична от нуля или, как говорят спектроскописты, переход является разрешённым. Молярный коэффициент поглощения при этих длинах волн или частотах достигает очень большой величины иногда 10 000 – 100 000 моль/л.см.
Обычно спектр выражают в виде графической зависимости оптической плотности А или молярного коэффициента поглощения ε от частоты ν или длины волны λ падающего света. Иногда удобно вместо А или λ использовать их логарифмы.
|
Распределение по частотам (или по длинам волн) значений молярного коэффициента поглощения называется спектром поглощения. |
На рис. (5.2) – (5.4) изображена полоса поглощения в координатах
lgε – ν и lgε – λ.
|
|
|
|
|
Рис. 5.2. Полоса поглощения |
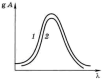
Рис. 5.3. Зависимость lgAотλ: 1 – раствор концентрации с в кювете толщиной l(см); 2 – раствор концентрации 1/4c или в кювете |
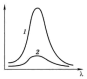
Рис. 5.4. Зависимость Аотλ 1 – раствор концентрации с в кювете толщиной l(см); 2 – раствор концентрации 1/4c или в кювете толщиной ¼(см) |
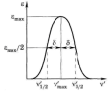
На рисунках
изображена отдельная полоса, реальный
спектр состоит из нескольких таких
перекрывающихся полос. На рис. 5.2 указаны
качественная характеристика
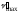 (частота
в максимуме поглощения) и количественная
–lgε).
Концентрация
вещества при соблюдении закона Бугера
– Ламберта - Бера пропорциональна
площади описываемой кривой в координатах
lgε
– ν, т. е.
(частота
в максимуме поглощения) и количественная
–lgε).
Концентрация
вещества при соблюдении закона Бугера
– Ламберта - Бера пропорциональна
площади описываемой кривой в координатах
lgε
– ν, т. е.
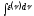 .
Вычисление этого интеграла при отсутствии
компьютеров представляло собой довольно
трудоёмкую задачу. В современных
приборах, оснащённых компьютерами, это
не представляет никаких проблем. Важной
аналитической характеристикой полосы,
которая используется для упрощённого
вычисления интеграла поглощения является
также полуширина полосы поглощения
.
Вычисление этого интеграла при отсутствии
компьютеров представляло собой довольно
трудоёмкую задачу. В современных
приборах, оснащённых компьютерами, это
не представляет никаких проблем. Важной
аналитической характеристикой полосы,
которая используется для упрощённого
вычисления интеграла поглощения является
также полуширина полосы поглощения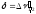 (рис. 5.2).
(рис. 5.2).
На рис. (5.3) и (5.4) показано как концентрация анализируемого раствора и ширина кюветы т. е. толщина светопоглощающего слоя влияют на общий вид спектра.
Чувствительность метода. При прочих равных условиях чувствительность метода абсорбционной спектроскопии определяется величиной молярного коэффициента поглощения. При очень больших ε метод может использоваться даже для определения следов веществ (менее 10-3%).
Фотометрические измерения. Фотометрией обычно называют электронную спектроскопию в видимой области, т. е. спектральное исследование окрашенных или изменяющих свою окраску в процессе реакций соединений. Фотометрические измерения возможны для таких соединений, у которых энергетические электронные уровни расположены настолько близко, что для возбуждения достаточно излучения с энергией видимой области спектра.
Оптимальные условия для фотометрического анализа. В уравнение основного закона светопоглощения входит концентрация окрашенного (в общем случае светопоглощающего) соединения, поэтому превращение определяемого компонента в такое соединение является важнейшей операцией фотометрии. Окрашенные соединения в растворе получаются в результате, главным образом, реакций окисления – восстановления и комлексообразования. Важно, чтобы такие реакции проходили полностью и до конца.
Для фотометрического определения важно выбрать аналитическую длину волны. Аналитическую длину волны обычно выбирают в максимуме полосы поглощения. Идеальной полосой для этой цели является, неперекрывающаяся с другими полосами, полоса высокой интенсивности. Например, при ε = 103 минимальная определяемая концентрация цветного соединения ~ 10-5 моль/л.
Толщина поглощающего слоя. Уравнение закона Бугера – Ламберта – Бера показывает, что чем больше толщина слоя, тем больше оптическая плотность и, следовательно, тем более чувствительным будет определение при прочих равных условиях. Однако с увеличением толщины слоя (длины оптического пути) возрастают потери на рассеяние света, особенно при работе с растворами. Кюветы с толщиной слоя больше, чем 5 см для фотометрии обычно не применяются.
Метод градуировочного графика. В соответствии с законом Бугера – Лаберта – Бера график в координатах оптическая плотность – концентрация должен быть линеен и прямая должна проходить через начало координат. Для построения такого графика достаточно, вообще говоря, одной экспериментальной точки. Однако, градуировочный график обычно строят не менее, чем по трём точкам, что повышает точность и надёжность определения. При нарушении линейной зависимости А от с, число точек на графике должно быть увеличено. Применение градуировочного графика является наиболее распространённым т точным методом фотометрических измерений. Основные ограничения связаны с трудностями приготовления эталонных растворов и учётом влияния так называемых третьих компонентов, т. е. компонентов, которые находятся в пробе, сами не определяются, но на результат влияют.
Метод молярного коэффициента поглощения. При работе по этому методу определяют оптическую плотность нескольких стандартных растворов (Аст), для каждого раствора рассчитывают ε = Аст/(lcст) и полученное значение ε усредняют. Затем измеряют оптическую плотность анализируемого раствора (Ах) и рассчитывают концентрацию сх по формуле
|
сх = Ах/(εl). |
Ограничением метода является обязательное подчинение анализируемой системы закону Бугера – Ламберта – Бера, по крайней мере, в области исследуемых концентраций.
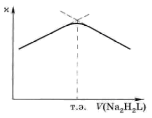
Фотометрическое титрование. В методе фотометрического титрования точка эквивалентности определяется с помощью фотометрических определений. Входе такого титрования изменяется светопоглощение раствора. Естественно, реализация этого метода возможна, если имеется подходящий индикатор или хотя бы один из компонентов поглощает свет в нужной области. Например, при титровании железа(II) дихроматом кривая фотометрического титрования имеет вид, изображённый на рис. 5.5. До точки эквивалентости (т. э.) оптическая плотность раствора практически не меняется, а после точки эквивалентности она линейно возрастает пропорционально объёму добавленного дихромата. Точку эквивалентности находят графически. Для этого достаточно иметь несколько точек, характеризующих недотитрованный раствор, и несколько точек для перетитрованного раствора.
|
|
|
Рис. 5.5. Кривая фотометрического титрования железа(II) дихроматом |
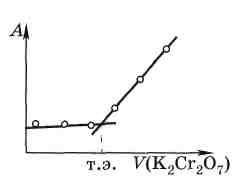
При титровании разбавленных или слабоокрашенных растворов кривая не имеет резкого минимума. Для нахождения точки эквивалентности в таких системах приходится применять более сложные графические построения или специальную математическую обработку.
Основным достоинством метода фотометрического титрования является возможность анализа слабоокрашенных и разбавленных растворов, которые часто невозможно оттитровать другими методами.
Электрохимические методы анализа. Потенциометрия и ионометрия. Потенциометрические методы основаны на измерении электродвижущих сил (ЭДС):
|
Е = Е1 – Е2, |
где Е – электродвижущая сила; Е1, Е2 – потенциалы электродов исследуемой цепи.
Потенциал электрода Е связан с активностью и концентрацией веществ, участвующих в электродном процессе, уравнением Нернста:
|
|
(5.3) |
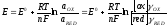 ,
,
где Е0 – стандартный потенциал редокс-системы; R – универсальная газовая постоянная, равная 8,312 Дж/(моль . К); Т – абсолютная температура, К; F – постоянная Фарадея, равная 96 485 Кл/моль; n – число электронов, принимающих участие в электродной реакции.; aOX, aRED – активности соотвественно окисленной и восстановленной форм редокс-системы; [ox], [red] – их концентрации γOX, γRED – коэффициенты активности (см. гл.3).
Интенсивное развитие потенциометрии в последние годы связано с появлением разнообразных типов ионоселективных электродов, позволяющих проводить прямые определения концентрации многих ионов в растворе, и успехами в конструировании и массовом выпуске приборов для потенциометрических измерений.
Потенциометрические методы анализа подразделяются на прямую потенциометрию (ионометрию) и потенциометрическое титрование. Методы прямой потенциометрии основаны на прямом применении уравнения Нернста (5,3) для нахождения активности или концентрации участника электродной реакции по экспериментально измеренной ЭДС цепи или потенциалу соответствующего электрода. При потенциометрическом титровании точку эквивалентности определяют по резкому изменению (скачку) потенциала вблизи точки эквивалентности.
Для проведения потенциометрического анализа обычно собирают гальванический элемент (см. гл. 3), на одном из электродов (индикаторном) протекает реакция с участием определяемого иона. При схематическом изображении различных гальванических элементов используют условную запись. Форма и символика схематического изображения гальванических элементов установлены решением ИЮПАК. По этим правилам формулы веществ, находящиеся в одном растворе, записывают через запятую, а границу между электродом и раствором или между разными растворами обозначают вертикальной чертой |. Двойная вертикальная черта || показывает, что так называемый диффузионный потенциал, возникающий на поверхности раздела растворов разного состава, сведён к минимуму или элиминирован с помощью солевого мостика. Так, например, гальванический элемент, состоящий из водородного и хлорсеребряного электродов, условно может быть изображён схемой Pt, (H2) | 0,1 M H2SO4 || 0,1 M KCl | AgCl, Ag. ЭДС такого элемента обычно измеряется компенсационным методом, когда ЭДС исследуемого элемета полностью компенсируется внешним источником напряжения.
Прямая потенциометрия (ионометрия). Наибольшее распространение среди прямых потенциометрических методов получил метод определения рН.
Для определения рН применяется уравнение (5.4), вывод которого мы приводить здесь не будем. Гальванический элемент, используемый для определения численного значения рН предложил Зёренсен в 1909г.
Pt, H2 | HCl, x || KCl; 0,1 M | Hg2Cl2, Hg
Уравнение (5.4) показывает, что для точного определения рН необходимы данные по диффузионным потенциалам и по активности иона Cl- в 0,1 М KCl. Ни одна из этих величин не может быть получена совершенно строго, в связи с чем найденная экспериментальная величина рН также не является вполне строгой. Эти трудности были преодолены путём введения соответствующего Государственного стандарта на шкалу рН.
|
|
(5.4) |
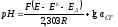
Принятая в России шкала рН основана на воспроизводимых значениях рН нескольких растворов.
Значения рН стандартных растворов устанавливаются путём измерения ЭДС цепей без переноса. Для этого чаще всего используют цепь типа
|
Pt(H2) | буферный раствор,КСl | AgCl,Ag |
В таких системах хотя и сохраняются трудности, связанные с оценкой коэффициентов активности отдельных ионов, но отпадает необходимость учёта диффузионного потенциала.
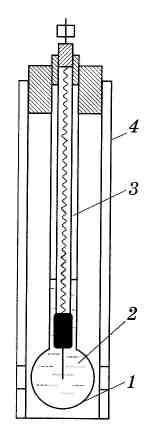
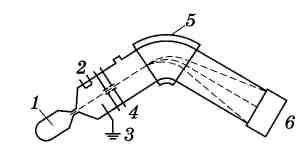
Стеклянный электрод. Для экспериментального определения рН и потенциометрического титрования наибольшее практическое применение нашёл стеклянный электрод, используемый в широком интервале рН и в присутствии окислителей.
Стеклянный электрод (рис. 5.6) представляет собой тонкостенный стеклянный шарик 1, заполненный раствором HCl или каким-либо буферным раствором 2. Внутрь шарика помещают хлорсеребряный электрод 3. Это устройство обычно закрывают защитной трубкой 4.
|
|
|
Рис. 5.6. Стеклянный электрод |
Схематически это устройство может быть представлено следующим образом:
|
Внешний раствор
|
Стеклянная мембрана |
Внутренний раствор
|
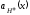
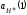
Не вдаваясь в технические подробности, отметим только, что потенциал такого электрода прямо пропорционален рН внешнего раствора, что выражается уравнением
|
|
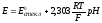 .
.
Стандартный
потенциал
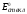 обычно
не определяют. При использовании
заводских рН-метров эта операция
заменяется настройкой приборов по
стандартным буферным растворам, так
как шкала рН-метров проградуирована
непосредственно в шкале рН.
обычно
не определяют. При использовании
заводских рН-метров эта операция
заменяется настройкой приборов по
стандартным буферным растворам, так
как шкала рН-метров проградуирована
непосредственно в шкале рН.
Кроме стеклянного электрода, который с успехом применяется для измерения рН, промышленность в настоящее время выпускает иoноселективные электроды для определения самых разных ионов – Li+, Na+, K+, Rb+, Cs+, Ag+, Tl+ и NH4+. У всех этих электродов принцип действия такой же, как и у стеклянного электрода, отличным является только состав стекла из которого сделаны мембраны.
Сульфидсеребряный электрод. Сульфидсеребряный электрод относится к твёрдым ионоселективным электродам. В таких электродах ионочувствительный элемент изготовляется из малорастворимого кристаллического вещества с ионным характером проводимости. Конструктивно такие электроды сходны со стеклянным: в обоих электродах мембрана разделяет исследуемый раствор и раствор сравнения, в котором находится электрод сравнения (обычно хлорсеребряный).
В сульфидсеребряном электроде, роль мембраны выполняют кристаллы Ag2S. Подвижными в этой мембране являются ионы Ag+. Серебро этим электродом может быть определено до концентрации 10-7моль/л. Примерно в таких же концентрациях могут быть определены и ионы S2-.
Плёночные электроды. В плёночных электродах роль мембраны играет специальная тонкая плёнка. У плёночных электродов такой же механизм действия, что и у мембранных, но они долговечнее и удобнее в работе.
Потенциометрическое титрование. Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала. Естественно это наблюдается только тогда, когда один из участников реакции титрования является и участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом, в качестве электрода сравнения обычно применяется хлорсеребряный электрод.
Кривая титрования получается в координатах: Е (обычно в милливольтах – мВ) – Vмл (объём добавленного титранта). Основное удобство при использовании потенциометрического титрования - отпадает необходимость в цветных индикаторах, что, конечно, повышает объективность анализа и полностью исключает субъективный фактор. Все расчёты по кривой титрования полностью аналогичны таковым в методах кислотно-основного титрования с индикаторами (см. гл. 4).
Кроме кислотно-основного титрования метод потенциометрии используется при комплексонометрическом и окислительно-восстановительном титрованиях с использованием, естественно, соответствующих электродов.
Основные правила работы с рН-электродами. Перед работой стеклянный электрод несколько суток вымачивают в 0Ю1 М растворе HCl. При этом ионы водорода из раствора обмениваются на ионы натрия из стеклянной мембраны и в системе устанавливается некоторое равновесие. Только подготовленный таким образом электрод, в котором протоны поверхности стекла находятся в равновесии с протонами раствора, может быть использован для определения рН.
Преимущества и недостатки метода. К преимуществам метода относится его доступность, простота работы, применимость в широкой области рН, быстрое установление равновесия и возможность определения рН в окислительно-восстановительных системах. Приборы для потенциометрии это серийно выпускаемые обычные потенциометры с соответствующей градуировкой (рН-метры, милливольтметры). Электроды также выпускаются промышленностью. Метод потенциометрии даёт воспроизводимые и надёжные результаты, в частности, потому что, как отмечалось выше, он исключает субъективный фактор в определении точки эквивалентности.
К недостаткам метода можно отнести хрупкость конструкции, в частности, стеклянных электродов и ненадёжность последних при работе в сильнокислых и сильнощелочных средах.
Электролиз и кулонометрия. Электролизом называют химическое разложение вещества под действием электрического тока. На катоде (отрицательно заряженном электроде) происходит восстановление:
|
Fe3+ + e- = Fe2+ |
|
|
|
Cu2+ + 2e- = Cu (к), |
а на аноде (положительно заряженном электроде) окисление:
|
2Cl- – 2e- = Cl2 (г)0 |
При электролизе растворов сульфатов, фосфатов и некоторых других солей на аноде происходит окисление не SO22-- или PO43--, а ОН- - ионов:
|
2ОН- – 2е- = 1/2O2 + H2O, |
поскольку ОН- легче отдаёт свои электроны, чем SO42- или PO43-, даже в кислом растворе. На аноде может происходить окисление не только анионов, но и катионов. Например, ионы Pb2+ образуют диоксид:
|
Pb2+ + 2H2O = PbO2 + 4H+ + 2e- |
Основные законы электролиза. Основные законы электролиза установлены Фарадеем.
|
Масса вещества, выделившаяся при электролизе, пропорциональна количеству электричества, прошедшего через раствор. |
|
|
|
При прохождении через раствор одного и того же количества электричества на электродах выделяется одно и то же количество вещества эквивалента. |
Эти законы выражаются формулой
|
|
(5.5) |
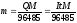 ,
,
где m – масса вещества, выделившегося при электролизе; Q – количество электричества; 96 485 – число Фарадея, равное количеству электричества, которое требуется для выделения молярной массы эквивалента вещества; I – сила тока; t – время электролиза.
Важной характеристикой процесса электролиза является выход по току, равный отношению количества выделившегося вещества к тому количеству вещества, которое должно было выделиться по закону Фарадея, т. е. в соответствии с уравнением (5.5).
Кулонометрия и кулонометрическое титрование. Принцип и теоретические основы кулонометрии были известны давно, однако широкое применение в аналитических методах она нашла лишь с конца 40-х годов.
|
В кулонометрических методах определяют количество электричества, которое расходуется в ходе электрохимической реакции. |
Различают два основных метода кулонометрических определений: прямую кулонометрию и кулонометрическое титрование. В методах прямой кулонометрии анализируемое вещество непосредственно подвергается электрохимическому превращению в кулонометрической ячейке и по измерению количества электричества определяют количество вещества эквивалента.
В методе кулонометрического титрования определяемое вещество реагирует с титрантом, который получается в кулонометрической ячейке при электролизе специально подобранного раствора.
В методе кулонометрического титрования используют установку с постоянной силой тока. Так как титрант генерируется в количестве, точно эквивалентном содержанию анализируемого вещества, то по количеству электричества, израсходованного на генерацию титранта можно рассчитать содержание определяемого вещества. Блок-схема установки для кулонометрического титрования приведена на рис. 5.7. Пульт-переключатель 4 питается током
|
|
|
Рис. 5.7. Блок-схема для кулонометрического титрования |
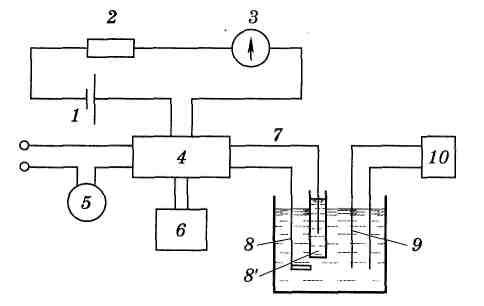
стабилизированного напряжения от аккумуляторной батареи 1 через сопротивление 2 и амперметр 3. Постоянство силы тока в генераторной цепи 7 контролируется потенциометром 6 по падению напряжения на стандартном сопротивлении. Пуск секундометра 5 и включение генераторной цепи 7 производится через пульт одновременно (8 и 8/ – генераторные электроды). Конец реакции фиксируется с помощью индикаторных электродов 9 измерительного потенциометра 10. Титрант генерируется в результате электролиза на электроде 8 (рабочий генераторный электрод). Вторым электродом схемы генерации является так называемый вспомогательный электрод 8 /. Его обычно изолируют от раствора анализируемого вещества, помещая в трубку с дном из пористого стекла, так как продукт реакции на вспомогательном электроде нередко мешает кулонометрическому определению. Индикаторными электродами могут быть два платиновых электрода, если для индикации применяется амперометрический метод, или платиновый и каломельный, если используется потенциометрическая индикация, и т. д. Может быть использован также спектрофотометрический или какой-либо другой способ определения точки эквивалентности.
Кулонометрическое титрование имеет некоторые преимущества перед обычными титриметрическими методами. Наиболее существенным достоинством кулонометрического титрования является то, что рабочий раствор в этом методе не готовят и не стандартизируют: титрант генерируется электрохимически непосредственно в присутствии анализируемого вещества и в количестве необходимым только для данного титрования.
Эквивалентная и удельная электропроводность. Перенос электричества в проводниках первого рода – металлах – осуществляется движением электронов по проводнику в направлении от отрицательного полюса источника тока к положительному. В проводниках второго рода – растворах электролитов – перенос электричества осуществляется движением ионов.
|
Электрической проводимостью называют способность вещества проводить электрический ток под действием внешнего электрического поля. Её единицей измерения является сименс (См). |
Закон Ома остаётся справедливым и для растворов электролитов:
|
|
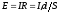 ,
,где Е– разность потенциалов между электродами, В;I– сила тока, А;R– сопротивление, Ом;ρ– удельное сопротивление Ом ∙ см;l– расстояние между электродами, см;S– сечение (площадь поверхности), см.
Величина а = 1/ρ, обратная удельному сопротивлению электролита, называется удельной электрической проводимостью.
|
Удельная электрическая проводимость равна электрической проводимости 1 см3 раствора, находящегося между параллельными электродами площадью 1 см2 при расстоянии между ними 1 см, или, другими словами, - это электрическая проводимость столба раствора длиной 1 см и площадью поперечного сечения 1 см2. Её единицей измерения является сименс на см - См/см. |
|
Эквивалентной электрической проводимостью называют проводимость раствора, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см. Её единицей измерения является См · см2/(моль экв). |
Кондуктометрия. Прямая кондуктометрия. Методы прямой кондуктометрии основаны на том, что в области разбавленных и умеренно концентрированных растворов электрическая проводимость растёт с увеличением концентрации электролита. Данные по электрической проводимости растворов применяют также для определения растворимости малорастворимых соединений. Кондуктометрические измерения широко используются для определения констант равновесия.
Кондуктометрическое титрование. Виды кривых титрования. В методах кондуктометрического титрования измеряют электрическую проводимость раствора после добавления небольших определённых порций титранта и находят точку эквивалентности графическим методом в координатах а – V титранта.
Типичные кривые кондуктометрического титрования для реакций комплексообразования приведены на рис. 5.8 и 5.9.
|
|
|
|
Рис. 5.8. Кривая кондуктометрического титрованияFe3+раствором ЭДТА |
Рис. 5.9. Кривая кондуктометрического титрования Са2+раствором ЭДТА в буферном растворе (рН ~ 10) |
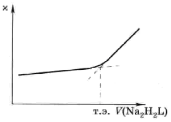
Прямое измерение электрической проводимости является наиболее эффективным методом контроля качества дистиллированной воды в лабораториях, технической воды в тонких химических и фармацевтических производствах. Методы прямой кондуктометрии используются для контроля качества молока, различных напитков и пищевых продуктов.
Кондуктометрическое титрование используется для определения сильных кислот и сильных оснований с высокой точностью (до 10-4 моль / л). Кривые кондуктометрического титрования ряда органических кислот (янтарной, адипиновой) имеют очень ярко выраженный излом при титровании слабым основанием.
Оба метода широко применяются для определения различных смесей.
Сущность и классификация хроматографических методов. Ионообменная хроматография. Хроматографию можно определить как, основанный на многократном повторении актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы вдоль неподвижного сорбента.
Сорбцией (от лат. – sorbeo – поглощаю) называют процесс поглощения твёрдым телом или жидкостью (сорбентом) газообразного или растворённого вещества (сорбата), обратный процесс называют десорбцией. Сорбацию подразделяют на адсорбцию – поглощение вещества (адсорбата) поверхностью твёрдого или жидкого адсорбента и абсорбцию − поглощение вещества (абсорбата) объёмом абсорбента.
Вещество подвижной фазы непрерывно вступает в контакт с новыми участками сорбента и частично сорбируется, а сорбированное вещество контактирует со свежими порциями подвижной фазы и частично десорбируется.
Различные методы хроматографии можно классифицировать по агрегатному состоянию фаз, способу их относительного перемещения, аппаратурному оформлению процесса и т. д. По агрегатному состоянию фаз хроматографические методы обычно классифицируют следующим образом (табл. 5.1).
Т а б л и ц а 5.1.* Классификация хроматографических методов по агрегатному состоянию фаз
|
Неподвижная фаза |
Подвижная фаза | |
|
газообразная |
жидкая | |
|
Твёрдая |
Газовая адсорбционная хроматография |
Жидкостная адсорбционная, ионообменная, ионная, тонкослойная, осадочная хроматография |
|
Жидкая |
Газожидкостная распределительная хроматография, капиллярная |
Жидкостная распределительная, высокоэффективная жидкостная, гель-хроматография |
По способу относительного перемещения фаз различают фронтальную, или элюэнтную, и вытеснительную хроматографию.
Ионообменная хроматография основана на обратимом стехиометрическом обмене ионов, находящихся в растворе, на ионы, входящие в состав ионообменника. Такой процесс называется ионным обменом. Иониты - ионообменные смолы или синтетические ионообменники. Наибольшее распространение в качестве синтетических ионообменников нашли органические ионообменные смолы, например, поперечно-сшитый полистирол, содержащий различные функциональные группы.
В зависимости от знака заряда функциональных групп ионообменные смолы являются катионитами или анионитами. Катиониты содержат кислотные функциональные группы [ – SO3-,– COO-, – PO3-, – N(CH2CO2-)], поэтому каркас катионита заряжен отрицательно.
Функциональными группами каркаса анионитов являются четвертичные – NR3+, третичные – NH2H+ или первичные – NH3+ аммониевые, пиридиновые или другие основания, а в качестве подвижных противоионов выступают анионы. Анионообменные смолы получаются также путём реакции полимеризации или поликонденсации с использованием различных аминосоединений (фенилендиамина, полиэтиленполиамина и т. п.), формальдегида и др.
Ионообменные методики характеризуются высокой селективностью или высокой избирательностью сорбции. Это объясняется тем фактом, что на место иона из смолы может встать только ион, обладающий сходными характеристиками, т. е. размером и зарядом, а сходные, ионы по химическим свойствам, могут очень сильно различаться, в частности, радиусом. Поэтому ионообменные методики применяются даже для разделения изотопов (14N и 15N).
Распределительная хроматография, её сущность. На твёрдый носитель наносится плёнка жидкой фазы и через колонку, наполненную таким сорбентом, пропускают жидкий раствор. Этот вид хроматографии и называют распределительной хроматографией. Жидкость, нанесённую на носитель, называют неподвижной жидкой фазой, а растворитель, передвигающийся через носитель, – подвижной жидкой фазой. Жидкостно-жидкостная хроматография может проводиться в колонке (колоночный вариант) и на бумаге (бумажная хроматография).
Разделение смеси веществ в распределительной хроматографии основано на различии коэффициентов распределения вещества между несмешивающимися жидкостями:
|
КП, Н = сП/сН, |
где сП исН– концентрация в подвижной и неподвижной фазах.
Колоночная распределительная хроматография. Адсорбенты с закреплённой на их поверхности жидкой фазой выпускаются промышленностью. Таким адсорбентом заполняется колонка. Эффективность колонки связана с вязкостью, коэффициентом диффузии и другими свойствами жидкостей. Носитель неподвижной фазы должен обладать достаточно развитой поверхностью, быть химически инертным, прочно удерживать на своей поверхности жидкую фазу и не растворятся в применяемых растворителях.
Бумажная распределительная хроматография. В бумажной распределительной хроматографии в качестве носителя используется специальная хроматографическая бумага.
Хроматографическая проба наносится на стартовую линию бумажной полоски и подвергается действию подвижной фазы (растворителя). Если компоненты окрашены, через некоторое время на хроматограмме можно увидеть отдельные цветные пятна.
Хроматографическая бумага должна быть химически чистой, нейтральной, инертной по отношению к компонентам раствора и подвижному растворителю и быть однородной по плотности. Имеют значение и некоторые другие свойства бумаги.
К растворителям обычно предъявляются следующие требования: растворители подвижной и неподвижной фаз не должны смешиваться, состав растворителя в процессе хроматографирования не должен изменяться, растворители должны легко удаляться с бумаги, быть недефицитными и безвредными для человека.
Основы газо-адсорбционной хроматографии. В газо-адсорбционной хроматографии неподвижная фаза – твёрдая, а подвижная – газообразная. В газо-адсорбционной хроматографии колонки заполняются твёрдым сорбентом. Адсорбция газа на твёрдом сорбенте подчиняется уравнению изотермы адсорбции:
|
|
(5.6) |
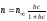 ,
,где n– количество адсорбированного вещества при равновесии;n∞- максимальное количество вещества, которое может быть адсорбировано на данном адсорбенте;b– постоянная;с– концентрация.
Зависимость количества поглощённого вещества от концентрации раствора или давления газа при постоянной температуре называют изотермой адсорбции. Уравнение (5.6) – это уравнение изотермы адсорбции Лэнгмюра.
В области небольших концентраций изотерма линейна. Действительно, при bc ‹‹ 1 знаменатель (5.6) становится равным единице, и уравнение (5.6) переходит в
|
n = n∞bc = Гс. |
(5.7) |
Это уравнение линейной адсорбции. Оно соответствует уравнению Генри (Г – коэффициент Генри). Область линейной адсорбции называют также областью Генри. Следует отметить, что количество адсорбированного вещества может определяться не только концентрацией, но и сродством адсорбанта к адсорбенту (специфичностью).
Основы газо-жидкостной хроматографии. Возможности хроматографического определения вещества в газовой фазе значительно возросли с открытием в 1952 г. метода газо-жидкостной хроматографии. При анализе по этому методу анализируемая газовая смесь проходит через колонку, наполненную твёрдым носителем, на поверхность которого нанесён тонкий слой жидкой фазы. Таким образом, с компонентами пробы здесь уже взаимодействует вещество жидкой плёнки.
В газо-жидкостной хроматографии вместо процесса адсорбции газа на твёрдом сорбенте имеет место процесс растворения газа в твёрдой плёнке, находящейся на твёрдом носителе. Различие в растворимости газов оказалось более существенным, чем различие в адсорбционных свойствах. Поэтому газо-жидкостная хроматография открыла более широкие возможности в анализе и разделении многокомпонентных смесей.
Эффективность разделения в газо-жидкостной хроматографии зависит главным образом от правильности выбора жидкой фазы. Строго обоснованных теоретических способов выбора жидкой фазы не существует. Однако требования к жидкой фазе предъявляются совершенно определённые. Жидкая фаза должна обладать высокой селективностью, быть химически инертной по отношению к компонентам смеси и твёрдому носителю, быть термически устойчивой, не растворять газ-носитель, иметь малую вязкость и быть не летучей.
В качестве жидкой фазы в газо-жидкостной хроматографии используются вазелиновое масло, силиконовое масло, фталаты (дибутил-, диоктил- и др.), диметилформамид, трикреазилфосфат, некоторые жидкие кристаллы и ряд других.
Газовый хроматограф. Независимо от сложности хроматографа основными узлами хроматографической установки являются дозатор (система ввода пробы), хроматографическая колонка и детектор. Кроме того, в установке имеются устройства для подачи газа-носителя, для преобразования импульса детектора в соответствующий сигнал и некоторые другие.
Дозатор предназначен для точного количественного отбора пробы и введения её в хроматографическую колонку. Газообразные и жидкие пробы вводятся с помощью специальных шприцов. Нередко в качестве дозатора в лабораторной практике используется обыкновенный медицинский шприц.
Хроматографические колонки весьма различны по форме, размерам и конструкционным материалам. Применяются прямые, спиральные и другие колонки длиной от 1 – 2 м и менее до нескольких десятков метров. Внутренний диаметр колонок обычно несколько миллиметров. Конструкционными материалами для колонок обычно служат медь, сталь, латунь, секло и др.
Большое влияние на процессы адсорбции-десорбции оказывает температура, поэтому практически все хроматографы имеют устройства для термостатирования.
Адсорбенты, которыми наполняют колонку, выпускаются промышленностью и имеют, как всякий товарный продукт, гарантии и инструкции по применению.
Детектор предназначен для обнаружения изменений в составе газа, прошедшего через колонку. Показания детектора обычно преобразуются в электрический сигнал и передаются фиксирующему или записывающему прибору, например на ленту электронного потенциометра. В современных приборах сигнал передаётся на компьютер.
Детекторы бывают по теплопроводности (катарометры), по плотности, по электрической проводимости, пламенные, пламенно-ионизационные и другие ионизационные детекторы, термохимические, пламенно-фотометрические и т. д. Детекторы выбирают в зависимости от свойств изучаемой системы.
Принцип работы, например, катарометра основан на измерении сопротивления нагретой платиновой нити, которое зависит от теплопроводности омывающего газа. Количество теплоты, отводимое от нагретой нити, зависит от состава газа. Чем больше теплопроводность определяемых компонентов смеси будет отличаться от теплопроводности газа-носителя, тем большей чувствительностью будет обладать катарометр.
В пламенно-ионизационных детекторах (ПИД) измеряют электрическую проводимость пламени водородной горелки. Чисто водородное пламя обладает очень низкой электрической проводимостью. При появлении в пламени органических соединений, происходит их ионизация и, как следствие повышение проводимости. ПИД позволяет обнаружить до 10-12 г вещества.
Качественный хроматографичекий анализ. Качественный состав пробы может быть установлен с помощью хроматографической методики по характеристике полученной хроматограммы. Типичная хроматограмма приведена на рис. 5.10. В идеальном случае каждому компоненту смеси отвечает свой пик.
|
|
|
Рис. 5.10. Хроматограммы смеси воды и кислот (1150С): 1 – вода; 2 – муравьиная кислота; 3 – уксусная кислота; 4 – пропионовая кислота; 5 – изомасляная кислота; 6 – н-масляная кислота; 7 – изовалерианова кислота |
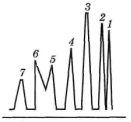
Собственно хроматографический качественный анализ основан на использовании характеристик удерживания – времени удерживания или пропорционального ему удерживаемого объёма и индексов удерживания. Для этой цели применяются относительные удерживаемые объёмы – формула (5.8). Идентификация исследуемых веществ проводится сравнением полученных и табличных данных.
|
Vr = trω, |
(5.8) |
где Vr – удерживаемый объём; tr – время удерживания; ω – объёмная скорость газа-носителя.
Иногда в смесь вводят вещество, наличие которого там предполагается (эталон) и сравнивают высоту или площадь пика до введения эталона и после.
Количественный хроматографический анализ основан на измерении различных параметров пика, зависящих от концентрации хроматографируемых веществ – высоты, ширины, площади и удерживаемого объёма – или произведения удерживаемого объёма на высоту. Выбор какого-либо параметра из перечисленных обусловлен конкретным видом хроматограммы. В частности, при достаточной стабильности условий хроматографирования и детектирования определяющим параметром пика можно считать его высоту.
Наиболее точным считается метод абсолютной калибровки. В этом методе экспериментально определяют зависимость высоты или площади пика от концентрации вещества, которое нужно определить в смеси и строят градуировочные графики, т. е. зависимость выбранного параметра пика от концентрации чистого определяемого вещества. Далее по этому графику находят концентрацию того же вещества, но уже в рабочей смеси.
Кинетические методы анализа. В кинетических методах анализа измеряемым свойством системы, на основании которого делают выводы о концентрации вещества, является скорость химической реакции. Пусть, например, вещества А и В реагируют между собой, образуя продукт реакции Х:
|
А + В = Х |
В начальный момент времени концентрации веществ А и В будут равны соответственно a и b. Концентрация продукта реакции в этот момент, естественно, будет равна нулю. В какой-то момент времени после начала реакции концентрация образующегося продукта Х будет равна х, а концентрации исходных веществ будут равны (а – х) и (b – x) соответственно. Скорость химической реакции при данной температуре, как известно пропорциональна произведению концентраций реагирующих веществ и нередко в степенях, отличных от единицы:
|
|
(5.9) |
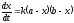 ,
,где k–константа скорости реакции.
Очень быстрые и очень медленные реакции в химико-аналитических целях являются малопригодными. Почти повсеместно принято считать, что аналитическая реакция (в кинетических методах её часто называют индикаторной реакцией) должна продолжаться не менее 1 мин и не более 2 ч. Оптимальным временем для измерения скорости аналитической реакции считается 10 – 15 мин. Наиболее часто в аналитических целях применяются так называемые каталитические реакции, скорость которых зависит от концентрации катализатора. При наличии в растворе катализатора в кинетическом уравнении (5.9) появляется соответствующий сомножитель:
|
|
(5.10) |
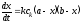 ,
,
где ск – концентрация катализатора.
Концентрацию одного из участников реакции, например вещества В, можно взять заведомо в большом избытке, так что его убыль в результате протекания реакции будет пренебрежимо равна и, следовательно, b – x ≈ b. Тогда, объединяя kb=а, вместо (5.10) получаем:
|
|
(5.11) |
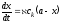 .
.
На рис. 5.11 приводятся типичные кинетические кривые. График показывает возрастание во времени концентрации вещества Х. Кривая 1 в начальный момент времени имеет линейный участок, т. е. в начальный момент
|
|
|
Рис. 5.11. Кинетические кривые |
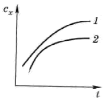
времени угловой коэффициент кинетической кривой постоянен. На кривой 2 линейный участок даже в начальный момент времени отсутствует.
Рассмотрим
кривую 1.
Уравнение (5.11) показывает, что величина
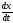 может быть постоянной только при условии
постоянстваа
– х, а это,
в свою очередь, будет возможно, если а
– х
может быть постоянной только при условии
постоянстваа
– х, а это,
в свою очередь, будет возможно, если а
– х
 а,
т. е. если концентрация вещества А в ходе
реакции существенно меняться не будет.
Кинетическое уравнение для этого
промежутка времени принимает вид:
а,
т. е. если концентрация вещества А в ходе
реакции существенно меняться не будет.
Кинетическое уравнение для этого
промежутка времени принимает вид:
|
|
(5.12) |
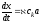 .
.
Это уравнение является основой различных вариантов кинетического метода, названных дифференциальными. Интегрирование даёт:
|
х = аскаt |
(5.13) |
Несколько
сложнее обработка данных, представленных
кривой 2.
Здесь нет области, в которой
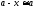 ,
поэтому приходится интегрировать
кинетическо уравнение (5.11). Разделив
переменные и проинтегрировав получаем
,
поэтому приходится интегрировать
кинетическо уравнение (5.11). Разделив
переменные и проинтегрировав получаем
|
|
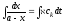
или
|
-ln(a – x) = аckt + const. |
Постоянную интегрирования находим из начальных условий при t = 0, x = 0 и, следовательно, -ln a = const. Окончательно можно записать:
|
|
(5.14) |
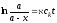 .
.
Методы анализа, основанные на применении этого уравнения, называют интегральными.
Основные приёмы кинетического анализа. Уравнения (5.12) – (5.14) в явном виде связывают кинетические характеристики реакции с концентрацией катализатора. Как видно, концентрация катализатора может быть найдена или непосредственно по скорости реакции, или по времени её протекания, или по концентрации образующихся продуктов. В зависимости от того, какое свойство или какая характеристика реакции используется для определения концентрации, выделяют методы тангенсов, фиксированной концентрации, фиксированного времени.
Кроме указанных существуют и другие методы, имеющие более частный характер: индукционного периода, непосредственного дифференцирования и ряд других.
В
качестве примера рассмотрим метод
тангенсов. В методе тангенсов измеряют
скорость индикаторной реакции обычно
по возрастанию одного из образующихся
веществ путём построения кинетической
кривой (рис. 5.11). На линейном участке
определяют тангенс угла наклон, т. е.
скорость
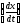 .
Уравнение (5.12) показывает, что в этом
случае скорость реакции пропорциональна
концентрации катализатора. Строят
градуировочный график в координатах
.
Уравнение (5.12) показывает, что в этом
случае скорость реакции пропорциональна
концентрации катализатора. Строят
градуировочный график в координатах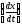 –ск.
–ск.
Далее
анализируют неизвестный раствор и
измеряют скорость реакции в тех же
условиях, в каких она измерялась для
построения градуировочного графика,
определяется тангенс угла наклона (tgα)
и по градуировочному графику находят
сх.
Использование кинетических методов анализа. Кинетические методы анализа характеризуются высокой чувствительностью и поэтому часто используются при определении малых и ультрамалых содержаний (до 10-8 –
– 10-6 мкг). Особенно эффективным оказалось применение кинетических методов для определения микропримесей в чистых и сверхчистых веществах и материалах.
В настоящее время известны десятки каталитических реакций, с помощью которых могут быть определены свыше 40 элементов периодической системы. Например, реакция иодида с пероксидом водорода без катализатора идёт очень медленно:
|
H2O2 + 2I- + 2H+ = I2 + 2H2O |
В присутствии следов молибдена, вольфрама, циркония, гафния, ниобия, тантала и других элементов она проходит за несколько минут. Скорость реакции легко определяется по возрастанию оптической плотности иодкрахмального раствора в единицу времени. Чувствительность индикаторной реакции достаточно велика. Например, с её помощью можно определить 0,01 мкг/л W, 0,02 мкг/л Мо, 0,1 мкг/л Zr и Hf.
С помощью кинетических методов анализа в большинстве случаев определяется не общая, а равновесная концентрация реагирующих веществ. В связи с этим кинетические методы успешно применяются для изучения различных равновесий в растворах (комплексообразование, кислотно-основное взаимодействие и др.).
Масс-спектральный анализ. Масс-спектральный анализ основан на способности газообразных ионов разделятся в магнитном поле в зависимости от отношения m/e, где m – масса, е – заряд иона. Ионизация молекул в газе происходит под действием потока электронов. Наиболее вероятными являются процессы образования однозарядных положительных ионов:
|
М + е- = М+ + 2е- |
Образование двух- и более высокозаряженных ионов, а также захват электрона с образованием отрицательных ионов являются менее вероятными процессами. По величине m/e определяют массовое число иона, а по интенсивности соответствующего сигнала судят о концентрации ионов.
Принципиальная схема масс-спектрометра приведена на рисунке 5.12.
|
|
|
Рис. 5.12. Принципиальная схема масс-спектрометра:1 – газообразная проба; 2 – катод; 3 – анод; 4 – ускоряющие пластинки; 5 – магнитное поле; 6 - детектор |
В камере анализируемое вещество переводится в газообразное состояние при давлении 10-2 – 10-3 Па. Далее молекулярный пучок ионизируется. Наибольшее распространение в аналитической практике получили приборы, в которых ионизация осуществляется электронной или ионной бомбардировкой либо искровым разрядом. Для ионизации, например, электронным ударом используется стабилизированный пучок электронов, перпендикулярный потоку пробы с энергией 10 – 100 эВ.
Образовавшиеся положительно заряженные ионы проходят через ускоряющие пластины, разность потенциалов между которыми достаточна высока (несколько тысяч вольт). Здесь они приобретают энергию eV , а их скорость возрастает до v. Энергия eV, очевидно, будет равна кинетической энергии ионов mv2/2, покидающий ионный источник со скоростью v:
|
eV = mv2/2 |
(5.15) |
После ускорения в электрическом поле ионы под прямым углом пересекают магнитное поле напряжённостью Н, подвергаясь, таким образом, действию силы Hev, направленной перпендикулярно движению иона. Поэтому траекторией движения ионов будет окружность радиуса r.
Приравнивая силы Hev = mv2/r, находим
|
|
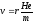 .
.
Подставляем эту величину в уравнение (5.15):
|
|
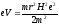 .
.
Отсюда получаем радиус окружности:
|
|
(5.16) |
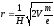 .
.
Ионы, описывающие дугу радиуса r, попадают в детектор. Детектирование ионов производится фотографическим или электрическим способом. При фотографическом детектировании пучок ионов попадает на фотопластинку, вызывая почернение пропорциональное числу ионов. В электрических детекторах масс-спектрометров измеряется ионный ток. В современных масс-спектрометрах информация из детектора передаётся на компьютер.
Перепишем уравнение (5.16) в виде
|
|
(5.17) |
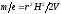 .
.
Меняя (сканируя) Н при постоянном V или меняя V при постоянном Н, можно направлять на детектор ионы с различным отношением m/e. Масс-спектр представляют зависимостью в виде спектрограммы или таблицы, содержащей величины m/e и соответствующие им интенсивности.
Качественный анализ. Масс-спектры многих веществ достаточно подробно изучены. Изданы специальные атласы и справочная литература. С помощью таких изданий и идентифицируются экспериментальные спектры.
Количественный анализ. Количественные измерения в масс-спектроскопии проводят по току, фиксируемому детектором. Пик ионного тока пропорционален содержанию компонента или его парциальному давлению:
|
I = kc = аp, |
где к, а – коэффициенты пропорциональности;с– концентрация;р– давление.
Практическое применение. Практическое применение масс-спектрометрии весьма разнообразно. Она нашла применение при изучении изотопного состава различных веществ, в органической химии для определения строения сложных молекулярных структур, в нефтехимии для анализа многокомпонентных веществ, для определения газов в металлах, в технологии неорганических соединений и т. д.
Экстракция в аналитической химии. При соприкосновении водного раствора вещества А с каким-либо неводным растворителем, не смешивающимся или ограниченно смешивающимся с водой растворённое вещество А будет распределяться между обоими растворителями и через некоторое время в такой системе установится равновесие
|
АВ ↔ А0, |
где АВ и А0 – вещество А в воде и в органическом растворителе соответственно.
Процесс переноса растворённого вещества из одной жидкой фазы в другую, с ней не смешивающуюся или ограниченно смешивающуюся жидкую фазу, называют жидкость-жидкостным распределением или распределением между двумя жидкостями.
Коэффициент распределения. Количественно этот процесс характеризуется законом распределения Нернста – Шилова, в соответствии с которым отношение концентраций растворённого вещества в обеих фазах при постоянной температуре постоянно и не зависит от общей концентрации растворённого вещества:
|
|
(5.18) |
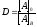 ,
,где D– коэффициент распределения; [A]0– аналитическая, т. е. суммарная концентрация всех форм вещества А в органической фазе; [A]B– то же, в водной фазе.
Величина D сохраняет своё постоянство только в отсутствии процессов диссоциации, ассоциации, полимеризации и других превращений растворённого вещества.
Константа
распределения. Отношение
концентрации (активности) вещества в
одной определённой форме (например,
МLn)
в фазе органического растворителя к
его концентрации (активности) в той же
форме в водной фазе называют константой
распределения
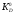 .
.
Коэффициент и константа распределения связаны с растворимостью вещества. В простейшем случае, когда вещество в обеих фазах существует в одной и той же форме (например, в виде недиссоциированных мономерных молекул), константа и коэффициент распределения равны отношению растворимостей вещества в органическом растворителе и воде.
Требования, предъявляемые к экстрагентам. Экстрагент – это органический растворитель, используемый для экстракции. Основные требования, предъявляемые к экстрагентам – по возможности низкая вязкость и невысокая плотность, низкая растворимость в воде, невысокая температура кипения (для того, чтобы можно было его легко отогнать для выделения экстрагируемого реагента), максимальная универсальность для того, чтобы можно было экстрагировать возможно большее количество веществ. Например, оксихинолин взаимодействует более чем с 50 элементами (Pd, Mo, W, V, Fe, Zr, Ga, In и многие другие). Экстрагент должен смешиваться с другими органическими растворителями, т. к. смешанные экстрагенты могут проявлять свойства, которые отсутствуют у индивидуальных экстрагентов. Например, селективность к какому-либо веществу. Иногда требуется, чтобы экстрагент смешивался с красителями или образовывал с экстрагируемыми соединениями окрашенные комплексы, тогда появляется возможность контроля путём фотокалометрии. Экстрагент должен быть не токсичен.
Механизмы и скорость экстракции. Распределение вещества между фазами является результатом многих физико-химических процессов, протекающих в обеих фазах и на границе между ними. Скорость экстракции определяется главным образом скоростью образования экстрагирующегося соединения и скоростью его распределения между фазами. Лимитирующей стадией в разных системах может быть как тот, так и другой процесс. Если скорость экстракции не зависит от интенсивности перемешивания, то лимитирующей стадией является скорость образования экстрагирующегося соединения. Например, скорость диссоциации на ионы и скорость образования ионных ассоциатов очень высоки и поэтому равновесие экстракции устанавливается быстро (3 – 5 мин).
Если экстрагирующееся соединение образуется в результате многостадийного химического процесса, то скорость экстракции, очевидно, будет определяться самой медленной стадией механизма химической реакции.
В простейшем случае, если экстрагируемое вещество представляет собой нейтральные мономерные молекулы и распределение вещества между фазами определяется исключительно растворимостью этого вещества, то скорость и механизм экстракции будет определяться свободной энергией сольватации молекул экстрагируемого вещества в каждой фазе и диффузионными процессами.
Основные количественные характеристики экстракции. Основными количественными характеристиками экстракции являются коэффициент и константа распределения (см. выше). Ещё одной важной количественной характеристикой экстракции является фактор (или степень) извлечения (R):
|
|
(5.19) |
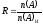 ,
,где n(A) – количество вещества в органической фазе;n(A)И– исходное его количество в водном растворе.
Фактор извлечения связан с коэффициентом распределения [D, см. (5.18)] и отношением объёмов водной и органической фаз (r = V0/VB) следующим образом:
|
|
(5.20) |
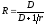 .
.
Уравнение (5.20) называется уравнением однократной экстракции. Уравнение многократной экстракции записывается следующим образом:
|
|
(5.21) |
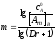 ,
,здесь
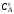 - концентрация вещества А в исходном
растворе;m – общее
число экстракций; [Am]В– концентрация вещества А в водной фазе
послеm-ой экстракции.
- концентрация вещества А в исходном
растворе;m – общее
число экстракций; [Am]В– концентрация вещества А в водной фазе
послеm-ой экстракции.
Если разделяются два вещества А и В, то качество экстракции характеризуется фактором обогащения S, показывающий, во сколько раз отношение количеств разделяемых веществ в фазе экстрагента превышает это отношение в исходном растворе до разделения:
|
|
(5.22) |
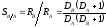 .
.
Опыт показывает, что в органическую фазу из водного раствора вещество переходит в форме электронейтральных частиц: координационных соединений, ионных ассоциатов и т. д. С увеличением температуры экстракция ухудшается.
Зависимость степени экстракции от коэффициента распределения и отношения объёмов органической и водной фаз. Из уравнения (5.20) очевидно, что степень экстракции (R) определяется двумя параметрами: коэффициентом распределения и отношением объёмов органической и водной фаз. Причём, чем больше коэффициент распределения, т. е. чем больше экстрагируемого вещества в условиях равновесия находится в органической фазе по сравнению с водной, тем больше степень извлечения и тем эффективней весь процесс в целом. Таким образом, зависимость пропорциональна.
С другой стороны R обратно пропорциональна величине отношения 1/r. Это означает, что чем меньше соотношение VB/V0, тем эффективней экстракция. Другими словами, при прочих равных условиях, органической фазы должно быть по возможности больше.
Перспективные физико-химические методы анализа и возможности их развития. Рассматривая перспективы развития физико-химического анализа, целесообразно выделить два направления: одно направление условно можно назвать «приборным» или «аппаратурным» и второе направление – это создание новых реактивов для анализа, его можно назвать «химическим» направлением.
Что касается первого направления, то с появлением доступных персональных компьютеров можно сказать, что эта отрасль преобразилась полностью или даже родилась во второй раз. В настоящее время трудно найти прибор, выпускаемый промышленностью, который не был бы оснащён компьютером. Это фактически произвело революцию в аналитической науке.
Во-первых, сократилось время обработки результатов, действительно, иногда на это требовались часы и даже сутки, с использованием компьютеров это время сократилось до минут и секунд, во-вторых, при наличии специальных программ появилась возможность обрабатывать слабые аналитические сигналы, за счёт методик отделения шумов, что реально повысило точность анализа без усложнения методик и приборной части и, в-третьих, компьютеризация позволила такие процедуры, как планирование эксперимента и статистическую обработку результатов сделать тривиальной для аналитического исследования.
Кроме, собственно, компьютеризации разрабатываются методики и материалы для физико-химического анализа, например, материалы для магнитов в ЯМР, ЭПР спектроскопии, для магнитов в приборах ион-циклотронного резонанса (ИЦР), материалы для сверхчувствительных ион-селективных электродов, датчиков и т. д.
В «химическом» направлении несомненным успехом является создание новых сверхселективных комплексонов и ионитов. С их помощью можно определять следовые количества многих веществ, особенно таких, которые даже в сверхмалых дозах способны оказывать решающее влияние на важные процессы, например, процессы жизнедеятельности.
Большим успехом химической науки можно считать синтез новых катализаторов и биологически активных соединений, которые применяются в аналитических целях.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
В.П. Васильев. Аналитическая химия. Т.I, II. М. – Дрофа. – 2007.
Ю.М. Глубоков, В.А. Головачёва, Ю.А. Ефимова. Аналитическая химия. М. – Академия. – 2006.
1AgClв растворе считается диссоциированным нацело.
**Строго говоря,аoxиaredдолжны быть возведены в степень, равную стехиометрическому коэффициенту этих веществ в уравнении полуреакций. Однако у большинства химико-аналитических полуреакций стехиометрические коэффициенты равны единице и только в некоторых отдельных случаях, таких какCr2O22-/2Cr3+, эти коэффициенты отличаются от единицы. В таких случаях, разумеется, стехиометрические коэффициенты должны быть учтены.
**Чтобы учесть изменение концентрации веществ при титровании за счёт увеличения объёма раствора, следует вычисленное значение концентрации умножить на коэффициент , гдеV– объём добавленного титранта.
**Раствор, содержащий все компоненты титруемого раствора, за исключением определяемого, и оттитрованный до соответствующего состояния, т. е. проявления аналитического эффекта в необходимой степени. Приготовленный «свидетель» используют в качестве образца сравнения. Окраска титруемого раствора и «свидетеля» должны быть одинаковы.
**Таблица имеет иллюстративный характер и некоторые виды хроматографии в неё не включены.