Гемофилия
А
67
Гемифациальная
атрофия прогрессирующая
Hemifacial
atrophy progressive MIM: 141300
Синоним:
синлром Парри Ромберга. Заболевание
описано в 1825 г. С. Parry Muuumil
/ьиые
диагностические признаки: атрофия
половины лица, неврологическая
симптоматика.
Клиническая
характеристики.
Типична атрофия кожи, подкожной жировой
клетча- ки. мышц, хрящей и костей
пораженной части лица. Атрофия
начинается с области глаза. затем
распространяется на бровь, угол рта.
шею; иногда вовлекается вся половина
тела Встречается витилнго. Патология
органов прения включает энофтальм.
паралич мышц глаза, птоз, лагофтальм.
гетерохро- мню радужки, синдром Горнера.
частые воспалительные заболевания,
иногда эти изменения отмечаются и
на непораженной стороне лица. На
пораженной стороне наблюдаются
деформация и уменьшение ушной раковины.
обнажение зубов, нарушение прикуса,
атрофия половины языка, тотальная
алопеция, выпадение ресниц и бровей,
задержка прорезывания зубов и разрушение
их корней. Неврологические изменения:
судорожные приступы по типу
джексоновской эпилепсии, невралгия
тройничного нерва, парестезии,
мигрень Заболевание начинается на I
2-м десятилетии жизни, медленно
прогрессирует в течение 3 лет, после
чего состояние стабилизируется.
Интеллект сохранен Соотношениептов—
Ml :Ж1 Тип
наследования
не установлен. Большинство случаев
спорадические. Предполагается
аутосомно-доминантное наследование
с неполной пенетрантностью.
Дифференциальный
диагноз
окуло-аури- куло-вертебральная дисплазия;
гемифациальная микросомия.
ЛИТЕРАТУРА
Lewkonia
R М
Lmery
R В
Progressive
hemifacial airophy (Parry Romberg syndrome) report with review
of genetics and nosology. —
Am.
J.
Med. Genet.. 1983.
v.
14.
p.
385—390.
NudlcnumK..
Andcrmnnn E'..
BerirundG
etal
The hemi 3syndrome:
hennhypenrophy, hcmiparesthesia. hemiareflexia and scoliosis. —
Brain.
1984.
v
107,
p
533
546.
Гемофилия
A
Hemophilia
A MIM: 306700
Синонимы
дефицит фактора VIII; классическая
гемофилия
Минимальные
диагностические притоки кровотечения,
гемартрозы; сниженная про- коагулянтная
активность фактора VIII.
Клиническим
характеристика
Заболевание распознается обычно на
2 -3-м году жизни, когда ребенок начинает
ходить, но в тяжелых случаях уже при
рождении наблюдаются кефалогематомы,
под- и внутрикож- ные кровоизлияния,
кровотечения из прочного канатика
Характерен гематомный тип кровоточивости.
Преобладают кровоизлияния в крупные
суставы, чаше всего в коленные.
локтевые и голеностопные
(рис. 37): подкожные,
внутримышечные и межмышечные
гематомы; гематурия; кровотечения при
травмах и хирургических вмешательствах,
в том числе при удалении зубов. Для
детей первых лет жизни характерны
кровотечения из травмированной слизистой
рта (травмы игрушками, прикусывание
языка). Геморрагии других локализаций
(гематомы внутренних органов,
кровоизлияния в мозг, желудочно-кишечные
кровотечения) встречаются реже
Гемартрозы приводят к остео- артрозам
и стойкой тугоподвижности суставов
При легких формах кровоточивость
проявляется только после массивных
травм или обширных оперативных
вмешательств. Отмечается снижение
прокоагулянтной активности фактора
VIII до 30 . (в тяжелых случаях — ниже 1%).
При определении частично» о тромбопласти
нового времени и в антнкоагулянтном
тесте выявляется гипо- коагуляция. При
тяжелой гемофилии отмечается
удлинение времени свертывания, времени
рекальцификацин цитратной плазмы,
снижение потребления протромбина
Популяциотшя
частота — I :
2500 живорожденных мальчиков.
Тип
наследования
— Х-сцепленный рецессивный. Ген
локализован на Xq28.
Дифференциальный
диагноз:
гемофилия В: болезнь Виллебранда.
ЛИТЕРАТУРА
Green
Р
М . Monlundon
A J Beniley D R Gi- anm lli F.
Genetics and molecular biology of haemophilias A and B. —
Blood
Coagulation Fibrinolysis. 1991.
v.
2.
p.
539
565.ViljtienD.. PearnJ. Brighton p Manifestations and natural history of idiopathic hemihypcrtrophy: a review of eleven cases. Clin Genet. 1984. V. 26. P. 81 —86.
68
Гемофилия
В
Рис.
37.
Гемофилия А. Гемартрозы коленных
суставов и стопы.
Гемофилия
В
Hemophilia
В MIM: 306900
Синонимы:
дефицит фактора IX; дефицит фактора
Кристмаса; болезнь Кристмаса; дефицит
плазменного тромбопластинового
компонента.
Клиническая
.характеристика.
Клинические проявления сходны с
таковыми при гемофилии А. При
определении частичного тромбопластинового
времени и в антикоагу- лянтном тесте
отмечается гипокоагуляция; удлиняется
время свертывания крови и время
рекальцификации цитратной плазмы,
снижается потребление протромбина.
Дефицит фактора IX можно косвенно
установить с помощью коррекционных
проб в тестах генерации тромбопластина,
образования тромбина и антикоагулянтном
тесте (в модификации Баркагана).
Достоверным считается снижение
активности фактора IX, определенное
количественным методом (ниже 30%).
Популяционная
частота —
примерно 1/10 от частоты гемофилии А.
Тип
наследования
- X сцепленный рецессивный. Ген
локализован на Xq27.l-q27.2.
Дифференциальный
дии *ноз:
гемофилия А; болезнь Виллебранда.
ЛИТЕРАТУРА
Green
P. Л/..
Montandon
4.
J.,
Bentley D. R.. G annelh F.
Genetics and molecular biology of haemophilias A and B. Blood
Coagulation Fibrinolysis. 1991.
v.
2.
p.
539
565.
Гемохроматоз
Hemochromatosis
MIM: 235200
Миншшльные
диигно* тича кие признаки: симптомы
портального цирроза печени и сахарного
диабета; бронзовая окраска кожи:
изменения на ЭКГ; повышение уровня
железа в сыворотке.
Ктиическая
.характеристика.
Гемохроматоз клинически проявляется
после 40 лет симптомами сахарного диабета
(60 80%); в 70% случаев диабет инсулинозависимын.
К другим возможным эндокринным
нарушениям относится дефицит гормонов
гипофи за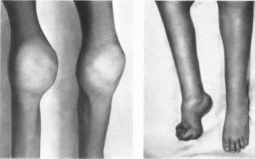
Гермафродитизм
истинный
69
(гонадотроиных,
адренокортикотропного и тиреотропного),
клинически проявляющийся генерали
юванной слабостью (45%). гипо- гонадизмом,
тестикулярной атрофией и снижением
либидо (15%). Постоянный симптом
гемохроматоза — гипертрофический
цирроз печени. Цвет кожи - от
металлического серого до бронзового
(90%). Встречаются жалобы на боли различной
локализации (30%), главным образом в
области печени. Поражение сердца
выявляется только на ЭК Г (н н зки й вол
ьтаж, у плошен ие и л и и нвер- сия зубца
Т. частичная блокада) У детей гемохроматоз
проявляется редко, при этом наблюдается
первичный инфантилизм; в юношеском
возрасте отмечается вторичная атрофия
гонад.
Тип
напсдования
— аутосомно-рецессив- ный. Ген локализован
на 6р21.3.
Дифференциальный
диагноз:
вторичный гемохроматоз (гемотрансфузии,
повышенное содержание железа в
пище).
ЛИТЕРАТУРА
Suildi
R .
Feingold
J
Idiopathic haemochromato- sis: an autosomal rcccssive disease. —
Clin.
Genet.. 1974.
v
5.
p.
234
241
Гепатолентикулярная
дегенерация
Hepatolenticular
degeneration MIM: 277900
Синонимы:
болезнь Вильсона Коновалова;
гепатоцеребральная дистрофия
Минимальные
диагностические признаки: снижение
концентрации церулоплазмина в плазме;
кольцо Кайзера Флейшера на радужке;
повышение содержания меди в печени;
гепатоспленомегалия; неврологические
нарушения.
Клииичсская
характеристики.
Заболевание проявляется в возрасте
от 6 до 50 лет. но наиболее часто — у
школьников. Первыми симптомами могут
быть гепатоспленомегалия. признаки
нарушения функции печени. ЦНС. иногда
почек; тромбоцитопения, лейкопения,
анемия. Поражение печени протекает
как подострын гепатит с желтухой,
рвотой. диспепсией; на поздних стадиях
развивается цирроз и портальная
гнпертензия. Неврологические изменения
включают днс- фагию. дизартрию,
слюнотечение, псевдо- бульбарные
симптомы, нарастающюю мышечную
ригидность, гиперкинезы. интенцн-
оннын
тремор. Отмечаются снижение интеллекта.
изменение поведения Патогномонич- ный
симптом — пигментное зелено-бурое
кольцо на радужке (кольцо Кай зера
Флейшера) Возможно изменение функций
почечных клубочков и канальцев
Биохимически выявляются гнпоцерулоплазминемия
(ниже 20 мг%), гипоальбумннемия, резкая
гипера- мннацидурия; повышен уровень
меди. На аутопсии обнаруживают отложенне
меди в мозге, печени, почках, селезенке,
роговице, радужной оболочке, хрусталике
глаза. Основной биохимический дефект
— дефицит церулоплазмина, обеспечивающего
транспорт меди в организме, что
приводит к повышению концентрации
меди в крови.
Преходящий
дефицит церулоплазмина встречается
при синдроме нарушенного всасывания
и в периоде новорожденное™, а также у
10% гетерозиготных носителей патологического
гена Концентрация меди в печени
повышается при атрезии желчного пузыря
и билиарном циррозе (уровень церулоплазмина
в этом случае нормальный).
Тип
нас илования
— аутосомно-рецессив- ный. Ген локализован
на I3ql4-q21.
Дифференциальный
диагноз:
синдром Пар- кинсона; хорея Гентингтона.
ЛИТЕРАТУРА
Сох
D..
Fraser
F.,
Sass-kortsak
A
A genetic study of Wilson's disease: evidence for heterogeneity. Am.
J. Hum. Genet., 1972,
v.
24,
p.
646
666.
Slovis
T . Dubois R. S..
Rodgerson
D U . Silverman A
The varied manifestations of Wilson's disease J. Pcdiatr.. 1971,
v.
78.
p.
548
584.
WahheJ
\1
Wilson's disease: yesterday, today and tomorrow. —
Mov.
Disord., 1988,
v.
3,
p.
10—29
Гермафродитизм
истинный
Hermaphroditism
true MIM 235660
Синоним:
амбисексуальность.
Минимальные
диагностические признаки: наличие
как мужских, так и женских гонад.
Клиническая
характеристика.
Основные проявления: двойственность
строения наружных половых органов
(чаше по мужскому типу с признаками
недостаточности ан- дрогенов —
гнпоспадией и расщеплением мошонки) и
смешанный характер вторичных половых
признаков. Диагноз ставят на основании
данных лапароскопии и гистологического
исследования гонад. Кариотип
70
Гиганттма
церебрального синдром
обычно
46.ХХ. реже — 46XY. 46.XX/46XY или
46.XX/47.XXY. Гонады могут
содержать одновременно тестикулярную
и овари- альную ткань (овотестис).
Различают гермафродитизм билатеральный
(с каждой стороны овотестис или яички
и яичник), унила- теральный (с одной
стороны нормальная гонада, с другой
- овотестис) и альтернативный (с одной
стороны яичко, с другой — яичник). Яичник
и овотестис расположены в брюшной
полости или паховом канале, яичко - в
мошонке или в паховом канале Чем больше
тестикулярной ткани, тем больше
вероятность опушения яичек в мошонку.
Гистологически строение яичников
нормальное, в яичках отсутствует
сперматогенез, имеется большое
количество клеток Лейдига.
Этиология:
I) мозаицизм 46.XX/46.XY или
46.XX/47.XXY; 2) транслокация
участка Y-хромосомы на
Х-хромосому или аутосо- му; 3) генная
мутация.
Риск
для сибсов ни
ЗКИЙ.
Тип
наследования
неизвестен.
Дифференциальный
диагноз:
различные формы мужского и женского
псевдогермафродитизма.
ЛИТЕРАТУРА
Berkovitz
G.
.-1.
True
hermaphroditism. J. Hopk. Med. J.. 1982.
v.
151.№6,
p.
290^
297.
Гигантизма
церебрального синдром
Cerebral
gigantism MIM: 117550
Синоним:
синдром Сотоса. Впервые описан в 1964 г.
J. F. Sotos Минимальные
диагностические признаки акромегалия,
усиленный рост, умственная отсталость,
нарушение координации.
Клиническая
характеристика.
При рождении вес и длина тела увеличены
(в среднем 3,9 кг и 55,2 см); в первые годы
жизни рост ускорен. Характерны
макроцефалия (100%). выступающие лобные
бугры (96%), увеличенные кисти и стопы
(рис. 38).
Костный возраст опережает паспортный.
Лицо гру
Рис.
38.
Синдром церебрального гиганпома
Увеличение кистей и с топ. высокий
выступающий лоб, прогения. гнпертелоризм.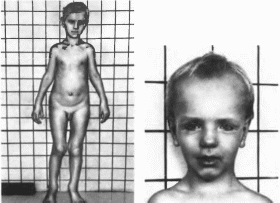
Гидроцефалия
71
бое.
с гипертелори
jmom
(91 %), антимонголо- идным разрезом
глаз, выступаюшей нижней челюстью
(83%). С возрастом лицо удлиняется,
усиливается прогения. Описаны гиперемия
лица с одутловатостью (лицевая плетора),
макроглоссия, высокое небо, косоглазие,
сколиоз, синдактилия стоп Степень
умственной отсталости варьирует,
но чаше бывает умеренной. Отмечаются
судороги, нарушение координации и
неспецифическне изменения на ЭКГ. При
пневмоэнцефало- графии выявляется
расширение желудочков мозга без
повышения внутричерепного давления
Синдром можно заподозрить уже при
рождении, но обычно диагноз ставится
в 2— 3 года. Продолжительность жизни не
изменена.
Тип
наследования
— аутосомно-доминант- ный. Большинство
случаев спорадические.
Дифференциальный
диагноз:
XYY-син-
дром;
синдром Беквнта Видемана; синдром
Вивера.
ЛИТЕРАТУРА
Cole
Т
R
Р
Hughes
Н
Е
Sotos
syndrome. J Med. Genet I990. v. 27.
p
57I—576
NevoS.
Lellzer M Bender!) A Lev) J
Evidence for autosomal recessive inheritance in cerebral
gigantism.—J Med Genet . 1974.
v
II. p I58 I65.
Zonana
J . Rirnoin D L Fisher D A.
Cerebral gigantism apparent dominant inheritance. Birth Defects,
I976. v. Xll<6). p 63—69.
Гидроцефалия
Hydrocephalus
MIM 236600. 307000
Минимальные
диагностические признаки увеличение
объема головы, расширение желудочков
мозга.
Рис.
39.
Гидроцефалия. Диспропорпионалыюе
увеличение мозговой части черепа,
нависающий лоб. косоглазие.