

69. Холестерин. Пути поступления, использования и выведения из организма. Уровень холестерина в сыворотке крови. Биосинтез холестерина, его этапы. Регуляция синтеза.
Холестерол
- стероид, характерный только для животных
организмов. Он синтезируется во многих
тканях человека, но основное место
синтеза - печень. В сутки в организме
синтезируется около 1 г холестерола; с
пищей поступает 300-500 мг. Холестерол
выполняет много функций: входит в состав
всех мембран клеток и влияет на их
свойства, служит исходным субстратом
в синтезе жёлчных кислот и стероидных
гормонов. Предшественники в метаболическом
пути синтеза холестерола превращаются
также в убихинон - компонент дыхательной
цепи и долихол, участвующий в синтезе
гликопротеинов. Холестерол за счёт
своей гидроксильной группы может
образовывать эфиры с жирными кислотами.
Этерифицированный холестерол преобладает
в крови и запасается в небольших
количествах в некоторых типах клеток,
использующих его как субстрат для
синтеза других веществ. Холестерол и
его эфиры - гидрофобные молекулы, поэтому
они транспортируются кровью только в
составе разных типов ЛП. Обмен холестерола
чрезвычайно сложен - только для его
синтеза необходимо осуществление около
100 последовательных реакций. Всего в
обмене холестерола участвует около 300
разных белков. Нарушения обмена
холестерола приводят к одному из наиболее
распространённых заболеваний -
атеросклерозу. Смертность от последствий
атеросклероза (инфаркт миокарда, инсульт)
лидирует в общей структуре смертности
населения. Атеросклероз - "полигенное
заболевание", т.е. в его развитии
участвуют многие факторы, важнейшие из
которых наследственные. Накопление
холестерола в организме приводит к
развитию и другого распространённого
заболевания - желчнокаменной болезни.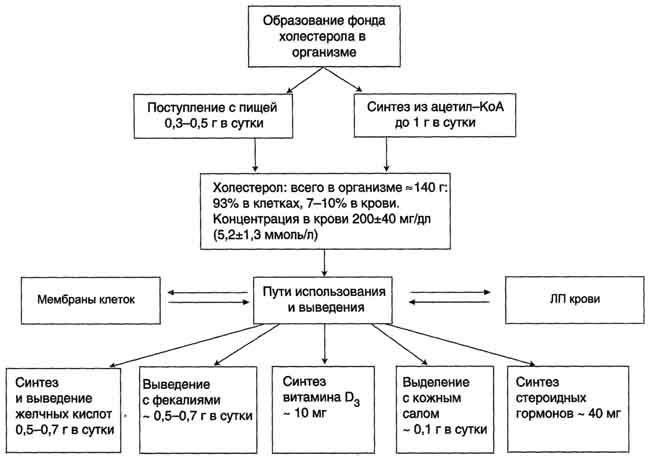
Реакции
синтеза холестерола происходят в
цитозоле клеток. Это один из самых
длинных метаболических путей в организме
человека.
Путь синтеза холестерола можно разделить на 3 этапа. Первый этап заканчивается образованием мевалоната (мевалоновой кислоты). На втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изопентенилпирофосфат. На третьем этапе синтеза холестерола сквален через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется 3 атома углерода, поэтому холестерол содержит 27 углеродных атомов.
Синтез холестерола. С5 - изопентенилпирофосфат; С1 - Фарнезилпирофосфат. Все атомы углерода холестерола происходят из ацетил-КоА. Сквален - углеводород линейной структуры - превращается ферментом циклазой в ланостерол, содержащий 4 конденсированных кольца и гидроксильную группу. Ланостерол через ряд последовательных реакций превращается в холестерол (I, II, III - этапы синтеза).
У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3.
Регуляция ключевого фермента синтеза холестерола (ГМГ-КоА-редуктазы) происходит разными способами.
Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы. При увеличении соотношения инеулин/глюкагон этот фермент дефосфорилируется и переходит в активное состояние. Действие инсулина осуществляется через 2 фермента:
фосфатазу киназы ГМГ-КоА-редуктазы, которая превращает киназу в неактивное дефосфорилированное состояние;
фосфатазу ГМГ-КоА-редуктазы путём превращения её в дефосфорилированное активное состояние. Результатом этих реакций служит образование дефосфорилированной активной формы ГМГ-КоА-редуктазы.
Следовательно, в абсорбтивный период синтез холестерола увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза холестерола - ацетил-КоА (в результате приёма пищи, содержащей углеводы и жиры, так как ацетил-КоА образуется при распаде глюкозы и жирных кислот).
Выведение холестерола из организма
Структурная основа холестерола - кольца циклопентанпергидрофенантрена - не может быть расщеплена до СО2 и воды, как другие органические компоненты, поступающие с пищей или синтезированные в организме. Поэтому основное количество холестерола выводится в виде жёлчных кислот.
Некоторое количество жёлчных кислот выделяется в неизменённом виде, а часть подвергается действию ферментов бактерий в кишечнике. Продукты их разрушения (в основном, вторичные жёлчные кислоты) выводятся из организма.
Часть молекул холестерола в кишечнике под действием ферментов бактерий восстанавливается по двойной связи в кольце В, в результате чего образуютря 2 типа молекул - холестанол и копростанол, выводимые с фекалиями. В сутки из организма выводится от 1,0 г до 1,3 г холестерола, основная часть удаляется с фекалиями.
Концентрация холестерола в крови взрослых людей составляет 200±50 мг/дл (5,2±1,2 ммоль/л) и, как правило, увеличивается с возрастом. Превышение нормальной концентрации холестерола в крови называют гиперхолестеролемией.
70. Роль липопротеинов низкой и высокой плотности (ЛПНП и ЛПВП) в обмене холестерина. Биохимические основы развития атеросклероза Количественное определение общего холестерина в сыворотке крови. Клиническое значение определения. Особенности липидного состава крови у детей.
Липиды являются в основе своей гидрофобными молекулами, они транспортируются в водной фазе крови в составе особых частиц – липопротеинов. Такие транспортные липопротеины можно сравнить с орехом, который имеет скорлупу и ядро. Поверхность липопротеиновой частицы ("скорлупа") гидрофильна и сформирована белками, фосфолипидами и свободным холестеролом. Триацилглицеролы и эфиры холестерола составляют гидрофобное ядро. Белки в липопротеинах обычно называются апобелками, выделяют несколько их типов – А, В, С, D, Е. В каждом классе липопротеинов находятся соответствующие ему апобелки, выполняющие структурную, ферментативную и кофакторную функции.
ЛП различаются по соотношению триацилглицеролов, холестерола и его эфиров, фосфолипидов и как сложные белки состоят из четырех классов.
хиломикроны (ХМ),
липопротеины очень низкой плотности (ЛПОНП, пре-β-липопротеины, пре-β-ЛП),
липопротеины низкой плотности (ЛПНП, β-липопротеины, β-ЛП),
липопротеины высокой плотности (ЛПВП, α-липопротеины, α-ЛП).
Хиломикроны и ЛПОНП ответственны, в первую очередь, за транспорт жирных кислот в составе ТАГ. ЛП высокой и низкой плотности – за транспорт холестерола и жирных кислот в составе эфиров ХС.
Транспорт триацилглицеролов от кишечника к тканям (экзогенные ТАГ) осуществляется в виде хиломикронов (ХМ), от печени к тканям (эндогенные ТАГ) – в виде липопротеинов очень низкой плотности.В транспорте ТАГ к тканям можно выделить последовательность следующих событий:
1.Образование незрелых первичных ХМ в кишечнике. 2.Движение первичных ХМ через лимфатические протоки в кровь.
3.Созревание ХМ в плазме крови – получение белков апоС-II и апоЕ от ЛПВП. 4.Взаимодействие с липопротеинлипазой (ЛПЛ) эндотелия, которая отщепляет жирные кислоты от ТАГ. Жирные кислоты переходят непосредственно в данную ткань или, связываясь с альбумином, разносятся по организму. В результате количество ТАГ в хиломикроне резко снижается и образуются остаточные ХМ. 5.Переход остаточных ХМ в гепатоциты и полный распад их структуры. 6.Синтез ТАГ в печени из пищевой глюкозы. Использование ТАГ, пришедших в составе остаточных ХМ. 7.Образование первичных ЛПОНП в печени.
8.Созревание ЛПОНП в плазме крови – получение белков апоС-II и апоЕ от ЛПВП. 9.Взаимодействие с липопротеинлипазой эндотелия и потеря большей части ТАГ. Образование остаточных ЛПОНП (по-другому липопротеины промежуточной плотности, ЛППП). 10.Остаточные ЛПОНП переходят в гепатоциты и полностью распадаются, либо остаются в плазме крови. После воздействия на них печеночной ТАГ-липазы в синусоидах печени ЛПОНП превращаются в ЛПНП.
ЛП очень низкой плотности (пребеталипопротеиды): -синтезируются в печени из эндогенных и экзогенных липидов,
-в их составе преобладают ТАГ, около 40% от массы составляют белок, фосфолипиды и холестерол (8% белка, 60% ТАГ, 6% ХС, 12% эфиров ХС, 14% фосфолипидов), -основным белком является апоВ-100, выполняющий структурную функцию,
-в норме концентрация 1,3-2,0 г/л, -слабо атерогенны.
Функция: Транспорт эндогенных и экзогенных ТАГ от печени в ткани, запасающие и использующие жиры, т.е. в те же ткани, что и хиломикроны (в сутки в печни обр-ся от 20 до 50 г жира на экспорт)
Метаболизм: 1. Первичные ЛПОНП образуются в печени из пищевых жиров, достигающих гепатоцитов с остаточными хиломикронами, и новосинтезированных из глюкозы жиров, содержат только апоВ-100; 2. В крови первичные ЛПОНП взаимодействуют с ЛПВП и приобретают от них апоС-II и апоЕ, образуя зрелые формы. 3. Аналогично хиломикронам, на эндотелии капилляров ряда тканей зрелые ЛПОНП подвергаются воздействию липопротеинлипазы, которая находится на поверхности кл эндотелия сосудов с образованием свободных жирных кислот и глицерина. Жирные кислоты перемещаются в клетки органов и используются как Е материал, либо остаются в плазме крови и в комплексе с альбумином разносятся с кровью в другие ткани. 4. Остаточные ЛПОНП (также называемые липопротеины промежуточной плотности, ЛППП)
либо эвакуируются в гепатоциты посредством эндоцитоза, связанного с рецепторами к апоЕ и апоВ-100-белкам, либо после воздействия на них печеночной ТАГ-липазы (только в сосудах печени) превращаются в следующий класс липопротеинов – липопротеины низкой плотности (ЛПНП).
ЛП низкой плотности (ЛПНП или beta-липопротеипы): Образуются в крови. Состоят из 25% белка и 75% липидов. Главным компонентом является холестерин (примерно50%) в виде эфиров с линолевой кислотой и фосфолипиды. У здоровых людей до 2/3 всего холестерина плазмы находится в составе ЛПНП. Они являются главным поставщиком холестерина в ткани. ЛПНП регулируют синтез холестерина de novo. Большинство ЛПНП являются продуктами расщепления ЛПОНП липопротеидлипазой. На клеточных мембранах имеются рецепторы для ЛПНП, они взаимодействуют с апопротеинами ЛПНП. После узнавания проникают в клетки путем эндоцитоза, там распадаются под действием ферментов гидролаз в лизосомах. Освободившийся холистерин идёт на построение мембран и метаболические нужды клеток. Функция: транспорт холестерина в ткани, в том числе в печени.
ЛП высокой плотности (ЛПВП или а-липопротеины): Состав: 50% белков, 25% фосфолипидов, 20% эфиров холестерина и очень мало триацилглицеринов. Образуются главным образом в печени. В поверхностном слое ЛПВПобразуют комплексы с ферментом лецитинхолестеролацилтрансферазой (ЛХАТ). С помощью этого фермента остаток ж к-ты переносится с лицитина на свободный холестерин ЛПВП, превращая его в эфир (холестерид) и лизофосфотидилхолин. Холестерид является гидрофобным соединением, поэтому перемещается в ядро ЛПВП. Т.о. он нагружается эфирами холестерина, увел-ся в размерах и из дисковидного превращается в частицу сферической формы – зрелый ЛПВП. Далее он транспортируется в печень, где холестерин исп-ся на синтез желчных к-т. ЛПВП, благодаря ЛХАТ, забирают холестерин от других липопротеидов и транспортируют его в печень, предотвращая накопление его в клетках. Концентрация ЛПВП меняется в зависимости от ритма питания. Их мах кол-во ч/з 4-5 часов после еды. Ч/з 10-12 часов хиломикронов 0%, ЛПОНП 15%, ЛПНП 60%, ЛПВП 25%.
ЛПОНП и ЛПНП считают атерогенными, то есть вызыва¬ющими атеросклероз. ЛПВП — антитиатерогенными.
Нарушения липидного обмена могут быть как первичными, так и вторичными, т.е. вызванными патологией эндокринной системы или компенсаторные при различных заболеваниях.
Нарушения переваривания и всасывания липидов сопровождаются развитием стеатореи (повышенное содержание липидов и жирных кислот в кале) и обусловливаются одной из следующих причин:
1.Дефицит панкреатической липазы, связанный с заболеваниями поджелудочной железы.
2.Дефицит желчи в кишечнике, обсуловленный заболеваниями печени или желчевыводящих путей.
3.Угнетение ферментных систем ресинтеза триглицеридов в стенке кишечника при его заболеваниях.
Повышение липопротеидов называется гиперлипопротеидемией. Главная опасность этого состояния связана с тем, что повыш-ся вер-ть возникновения атеросклероза. Вер-ть заболевания тем выше, чем больше отношение ЛПНП к ЛПВП в крови.
Атеросклероз -это патология, которая характеризуется отложением, главным образом, холестерина в стенке крупных сосудов (аорта, коронарные сосуды, сосуды мозга и т.д.) с образованием вначале пятен, полосок. Затем на их месте образуются утолщения (атеросклеротические бляшки). Эти липидные бляшки являются сво¬еобразным инородным телом, вокруг которого развивается со¬единительная ткань, затем наступает кальцификация пораженного участка сосуда. Сосуды становятся неэластичными, плотными, ухудшается кровоснабжение ткани, а на месте бляшек могут возникать тромбы. В стенке сосудов есть два защитных механизма от избыточного отложения холестерина: Работа липопротеидлипазы, которая расщепляет жир липопротеидов, делает их меньше по размеру;ЛПВП, которые уносят холестерин.Ожирение: У нормально упитанного человека жиры составляют около 15% массы тела. При сбалансированном питании количество жира в организме не изменяется. При этом жиры жировой ткани все время обновляются, то есть одновременно идут липолиз и липогенез с равными скоростями. В результате жиры жировой ткани за несколько дней обновляются полностью. При длительном голодании и физических нагрузках липолиз идет с большей скоростью, чем липогенез. В результате количество депонированного жира уменьшается. Если липогенез опережает липолиз, наступает ожирение. Наиболее частой причиной ожирения является несоответствие между количеством потребляемой пищи и энергетическими тратами организма. Такое несоответствие возникает при переедании (особенно углеводов, так как они легко переходят в жиры), при гиподинамии (при этом мобилизация жира идет с более низкой скоростью) и, особенно, при сочетании этих факторов. Кроме того, ожирение является характерным признаком многих эндокринных заболеваний. Генетические заболевания, при которых происходит неполное расщепление полимерных веществ и их накопление, получили название лизосомные болезни накопления, так как они обусловлены дефектами специфических лизосомальных гидролаз. В случае накопления липидов такие болезни называются липидозы. При липидозах нарушается нормальный катаболизм липидов до соответствующих мономеров. Болезнь Вольмана – редкое аутосомно-рецессивное заболевание из-за дефекта кислой эстеразы лизосом, что обусловливает накопление эфиров холестерола в лизосомах печени, селезенки, надпочечников, костного мозга и тонкого кишечника. Проявляется в первые недели жизни рвотой, диареей и стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечников. Больные умирают в возрасте до 6 мес. Болезнь Шюллера-Кристиана аутосомно-рецессивное заболевание характеризуется отложением в плоских костях, твердой мозговой оболочке и коже холестерола и его эфиров. Характерными являются деструктивные изменения в костях, остеопороз, мозжечковые расстройства. Заболевание обычно начинается в возрасте до 10 лет, реже позднее. Мужчины болеют в 2 раза чаще, чем женщины. Течение заболевания прогрессирующее. Дефектный фермент неизвестен. Болезнь Гоше – отложение цереброзидов в макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Возникает в связи с аутосомно-рецессивным отсутствием гликоцереброзидазы (β-глюкозидазы). Основными симптомами заболевания являются спленомегалия, увеличение печени и селезенки, а также изменения в костях, проявляющиеся в виде остеопороза. При болезни Нимана-Пика наблюдается отложение сфингомиелина в клетках различных органов из-за дефицита сфингомиелиназы. Болезнь наследуется аутосомно-рецессивно, проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет. Болезнь Тея-Сакса (амавротическая семейная идиотия) является результатом дефекта N-ацетилгексозаминидазы, при котором происходит отложение ганглиозидов в клетках головного мозга, что сопровождается атрофией зрительных нервов, слепотой, слабоумием и смертью в младенческом возрасте.
Содержание липидных фракций новорожденных отличается от спектра этих веществ у более старших детей и взрослых тем, что у них значительно увеличено содержание альфа-липопротеинов и понижено количество.
71. Общая схема источников поступления и путей расходования аминокислот в тканях. Динамическое состояние белков в организме. Причины необходимости постоянного обновления белков организма. «Незаменимые» аминокислоты.
АК, класс органических соединений, объединяющих в себе свойства кислот и аминов, т.е. содержащих наряду с карбоксильной группой - COOH аминогруппу - NH2. АК играют важную роль в жизни организмов, т.к. все белковые вещества построены из аминокислот. Все белки при полном гидролизе (расщеплении с присоединением воды) распадаются до свободных аминокислот, играющих роль мономеров в полимерной белковой молекуле. Источники АК в клетке – поступление с пищей, из крови, распад собственных внутриклеточных белков и синтез заменимых аминокислот, кетокислот. Основным экзогенными источником аминокислот являются белки пищи. Белки переводятся в доступную для организма форму при переваривании под действием протеолитических ферментов, входящих в состав желудочно-кишечных секретов. Свободные аминокислоты всасываются и после транспорта кровью включаются в клетках в различные пути использования. Путь дальнейшего превращения аминокислот зависит от вида и функции клетки, условий ее существования и гормональных влияний. Спектр веществ, получаемых клеткой из аминокислот, чрезвычайно широк.
Главным путь испол-я - синтез собственных белков. АК используются для синтеза других азотсодержащих соединений, например таких, как тироксин, адреналин, гистамин, выполняющих специфические функции. Аминокислоты используются также как источники энергии, включаясь в путь катаболизма. Многие растения и бактерии могут синтезировать все необходимые им аминокислоты из простых неорганических соединений. Большинство аминокислот синтезируются в теле человека и животных из обычных безазотистых продуктов обмена веществ и усвояемого азота. Однако восемь аминокислот (валин, изолейцин, лейцин, лизин, фенилаланин, метионин, треонин, триптофан) являются незаменимыми, т.е. не могут синтезироваться в организме животных и человека, и должны доставляться с пищей. Суточная потребность взрослого человека в каждой из незаменимых аминокислот составляет в среднем около 1 грамма. При недостатке этих аминокислот (чаще триптофана, лизина, метионина) или в случае отсутствия в пище хотя бы одной из них невозможен синтез белков и многих других биологически важных веществ, необходимых для жизни.
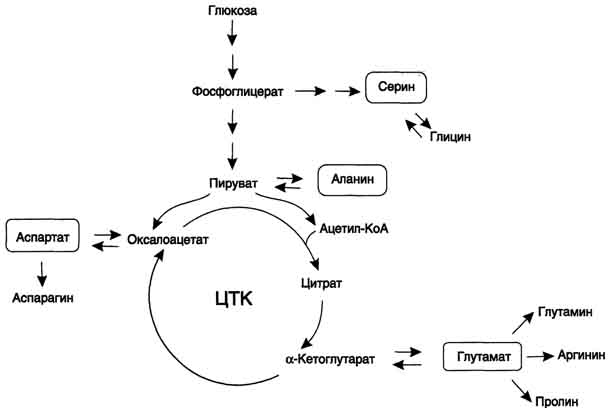
В организме человека возможен синтез восьми заменимых аминокислот: Ала, Асп, Асн, Сер, Гли, Глу, Глн, Про. Углеродный скелет этих аминокислот образуется из глюкозы. α-аминогруппа вводится в соответствующие α-кетокислоты в результате реакций трансаминирования. Универсальным донором α-аминогруппы служит глутамат. Путём трансаминирования α-кетокислот, образующихся из глюкозы, синтезируются аминокислоты. Глутамат также образуется при восстановительном аминировании α-кетоглутарата глутаматдегидрогеназой. Эти реакции обратимы и играют большую роль как в процессе синтеза аминокислот, так и при их катаболизме. Такие реакции, выполняющие двойную функцию, называют амфиболическими.
Бднако безбелковое питание (особенно продолжительное) вызывает серьёзные нарушения обмена и неизбежно заканчивается гибелью организма. Исключение даже одной незаменимой аминокислоты из пищевого рациона ведёт к неполному усвоению других аминокислот и сопровождается развитием отрицательного азотистого баланса, истощением, остановкой роста и нарушениями функций нервной системы.
Конкретные проявления недостаточности одной из аминокислот были выявлены у крыс, которым скармливали белки, лишённые определённой аминокислоты. Так, при отсутствии цистеина (или цистина) возникал острый некроз печени, гистидина - катаракта; отсутствие метионина приводило к анемии, ожирению и циррозу печени, облысению и геморрагии в почках. Исключение лизина из рациона молодых крыс сопровождалось анемией и внезапной гибелью (этот синдром отсутствовал у взрослых животных).
Недостаточность белкового питания приводит к заболеванию, получившему в Центральной Африке название "квашиоркор", что в переводе означает "золотой (или красный) мальчик". Заболевание развивается у детей, которые лишены молока и других животных белков, а питаются исключительно растительной пищей, включающей бананы, таро, просо и, чаще всего, кукурузу. Квашиоркор характеризуется задержкой роста, анемией, гипопротеинемией (часто сопровождающейся отёками), жировым перерождением печени. У лиц негроидной расы волосы приобретают красно-коричневый оттенок. Часто это заболевание сопровождается атрофией клеток поджелудочной железы. В результате нарушается секреция панкреатических ферментов и не усваивается даже то небольшое количество белков, которое поступает с пищей. Происходит поражение почек, вследствие чего резко увеличивается экскреция свободных аминокислот с мочой. Без лечения смертность детей составляет 50-90%. Даже если дети выживают, длительная недостаточность белка приводит к необратимым нарушениям не только физиологических функций, но и умственных способностей. Заболевание исчезает при своевременном переводе больного на богатую белком диету, включающую большие количества мясных и молочных продуктов. Один из путей решения проблемы - добавление в пищу препаратов лизина.


72. Катаболизм аминокислот. Общие пути распада аминокислот. Трансаминирование аминокислот. Схема реакций, ферменты, роль витамина В6. Биологическое значение трансаминирования. Диагностическое значение определения трансаминаз в сыворотке крови.
Непрямое
окислительное дезаминирование
(трансдезаминирование). Непрямое
окислительное дезаминирование включает
2 этапа и активно идет во всех клетках
организма.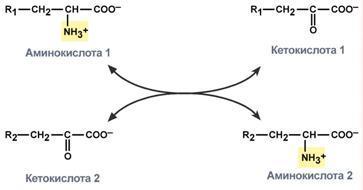
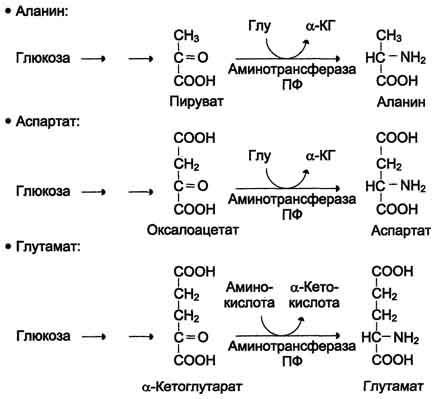
Первый этап заключается в обратимом переносе NH2-группы с аминокислоты на кетокислоту с образованием новой аминокислоты и новой кетокислоты – этот перенос называется трансаминирование и его механизм довольно сложен. В качестве кетокислоты-акцептора ("кетокислота 2") в организме обычно используется α-кетоглутаровая кислота, которая превращается в глутамат ("аминокислота 2"). В результате трансаминирования свободные аминокислоты теряют α-NH2-группы и превращаются в соответствующие кетокислоты. Далее их кетоскелет катаболизирует специфическими путями и вовлекается в цикл трикарбоновых кислот и тканевое дыхание, где сгорает до СО2 и Н2О. При необходимости (например, голодание) углеродный скелет глюкогенных аминокислот может использоваться для синтеза глюкозы в глюконеогенезе.
Второй
этап
состоит в отщеплении аминогруппы от
аминокислоты 2 – дезаминирование. В
организме человека дезаминированию
подвергается только глутаминовая
кислота. Второй этап осуществляется
глутаматдегидрогеназой (перейти вверх).
В организме коллектором всех аминокислотных
аминогрупп является глутаминовая
кислота, и только она подвергается
окислительному дезаминированию с
образованием аммиака и α-кетоглутаровой
кислоты. Фермент глутаматдегидрогеназа
имеется в митохондриях всех клеток
организма, кроме мышечных. Учитывая
тесную связь обоих этапов, непрямое
окислительное дезаминирование называют
трансдезаминирование. Если реакция
идет в митохондриях печени, аммиак
используется для синтеза мочевины,
которая в дальнейшем удаляется с мочой.
В эпителии канальцев почек реакция
необходима для удаления аммиака в
процессе аммониегенеза. Т.к. НАДН
используется в дыхательной цепи и
α-кетоглутарат вовлекается в реакции
ЦТК, то реакция активируется при дефиците
энергии и ингибируется избытком АТФ и
НАДН.
Роль трансаминирования и трансдезаминирования:
Реакции трансаминирования:
-активируются в печени, мышцах и других органах при поступлении в клетку избыточного количества тех или иных аминокислот – с целью оптимизации их соотношения,
-обеспечивают синтез заменимых аминокислот в клетке при наличии их углеродного скелета (кетоаналога),
-начинаются при прекращении использования аминокислот на синтез азотсодержащих соединений (белков, креатина, фосфолипидов, пуриновых и пиримидиновых оснований) – с целью дальнейшего катаболизма их безазотистого остатка и выработки энергии,
-необходимы при внутриклеточном голодании, например, при гипогликемиях различного генеза – для использования безазотистого остатка аминокислот в печени для кетогенеза и глюконеогенеза, в других органах – для его прямого вовлечения в реакции цикла трикарбоновых кислот.
-при патологиях (сахарный диабет, гиперкортицизм) обуславливают наличие субстратов для -глюконеогенеза и способствуют патологической гипергликемии.
Продукт трансаминирования глутаминовая кислота:
-является одной из транспортных форм аминного азота в гепатоциты,
-способна реагировать со свободным аммиаком, обезвреживая его.
Процесс
трансдезаминирования идет в организме
непрерывно, потому что сопряженные
реакции трансаминирования и дезаминирования
создают поток лишнего аминного азота
из периферических клеток в печень для
синтеза мочевины и в почки для синтеза
аммонийных солей.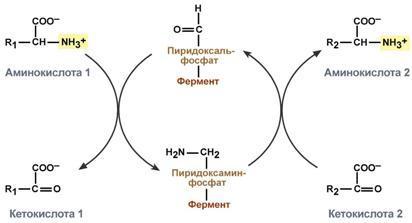
Механизм реакции трансаминирования непрост и протекает по типу "пинг-понг". Катализируют реакцию ферменты аминотрансферазы, Они являются сложными ферментами, в качестве кофермента имеют пиридоксальфосфат (активная форма витамина В6). В тканях насчитывают около 10 аминотрансфераз, обладающие групповой специфичностью и вовлекающие в реакции все аминокислоты, кроме пролина, лизина, треонина, которые не подвергаются трансаминированию.
Весь перенос аминогруппы совершается в две стадии:
1.к пиридоксальфосфату сначала присоединяется первая аминокислота, отдает аминогруппу, превращается в кетокислоту и отделяется. Аминогруппа при этом переходит на кофермент и образуется пиридоксаминфосфат.
2. на второй стадии к пиридоксаминфосфату присоединяется другая кетокислота, получает аминогруппу, образуется новая аминокислота и пиридоксальфосфат регенерирует.
Роль
и превращение пиридоксальфосфата
сводится к образованию промежуточных
соединений – шиффовых оснований
(альдимин и кетимин). В первой реакции
после отщепления воды образуется
иминовая связь между остатком аминокислоты
и пиридоксальфосфатом. Полученное
соединение называется альдимин.
Перемещение двойной связи приводит к
образованию кетимина, который гидролизуется
водой по месту двойной связи. От фермента
отщепляется готовый продукт – кетокислота.

После
отщепления кетокислоты к комплексу
пиридоксамин-фермент присоединяется
новая кетокислота и процесс идет в
обратном порядке: образуется кетимин,
затем альдимин, после чего отделяется
новая аминокислота. Чаще всего аминокислоты
взаимодействуют со следующими
кетокислотами: пировиноградной с
образованием аланина, щавелевоуксусной
с образованием аспартата, α-кетоглутаровой
с образованием глутамата. Однако аланин
и аспартат в дальнейшем все равно
передают свою аминогруппу на
α-кетоглутаровую кислоту. Таким образом,
в тканях осуществляется поток избыточных
аминогрупп на один общий акцептор –
α-кетоглутаровую кислоту. В итоге
образуется большое количество глутаминовой
кислоты. Далее глутаминовая кислота
может вовлекается в процессы связывания
аммиака (синтез глутамина) либо в прямое
окислительное дезаминирование.
В медицине нашло практическое применение определение активности двух ферментов трансаминирования – аланинаминотрансферазы (АЛТ, АлАТ) и аспартатаминтрансферазы (АСТ).
Оба фермента обратимо взаимодействуют с α-кетоглутаровой кислотой и переносят на нее аминогруппы от соответствующих аминокислот с образованием глутаминовой кислоты и кетокислот. Хотя активность обоих ферментов значительно возрастает при заболеваниях сердечной мышцы и печени, при поражении клеток миокарда наибольшая активность в сыворотке крови обнаруживается для АСТ, при гепатитах – для АЛТ.
73. Дезаминирование аминокислот: прямое, непрямое. Виды прямого дезаминирования. Окислительное дезаминирование. Оксидазы L-аминокислот. Глутаматдегидрогеназа. Схема реакции, кофактор, регуляция процесса.
Дезаминирование аминокислот - реакция отщепления α-аминогруппы от аминокислоты, в результате чего образуется соответствующая α-кетокислота (безазотистый остаток) и выделяется молекула аммиака.
Прямое дезаминирование – отщепление аминогруппы в виде аммиака. Аммиак выделяется в кровь и очень токсичен (этому виду дезаминирования подвергаются все аминокислоты, кроме лизина). Это не главный путь дезаминирования.
Виды прямого дезаминирования:
окислительное
восстановительное
гидролитическое
внутримолекулярное
Окислительное
дезаминирование. Наиболее
активно в тканях происходит дезаминирование
глутаминовой кислоты. Реакцию катализирует
фермент глутаматдегидрогеназа, коферментом
глутаматдегидрогеназы является NAD+.
Реакция идёт в 2 этапа. Вначале происходит
ферментативное дегидрирование глутамата
и образование а-иминоглутарата, затем
- неферментативное гидролитическое
отщепление иминогруппы в виде аммиака,
в результате чего образуется а-кетоглутарат.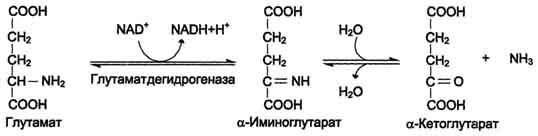

Окислительное дезаминирование глутамата - обратимая реакция и при повышении концентрации аммиака в клетке может протекать в обратном направлении, как восстановительное шинирование α-кетоглутарата.
Глутаматдегидрогеназа очень активна в митохондриях клеток практически всех органов, кроме мышц. Этот фермент - олигомер, состоящий из 6 субъединиц (молекулярная масса 312 кД). Глутаматдегидрогеназа играет важную роль, так как является регуляторным ферментом аминокислотного обмена. Аллостерические ингибиторы глутаматдегидрогеназы (АТФ, ГТФ, NADH) вызывают диссоциацию фермента и потерю глутаматдегидрогеназной активности. Высокие концентрации АДф активируют фермент. Таким образом, низкий энергетический уровень в клетках стимулирует разрушение аминокислот и образованиеα-кетоглутарата, поступающего в ЦТК как энергетический субстрат. Глутаматдегидрогеназа может индуцироваться стероидными гормонами (кортизолом).
Оксидаза L-аминокислот. В печени и почках обнаружен фермент оксидаза L-аминокислот, способный дезаминировать некоторые L-аминокислоты.
Коферментом в данной реакции выступает FMN. Однако вклад оксидазы L-аминокислот в дезаминирование, очевидно, незначителен, так как оптимум её действия лежит в щелочной среде (рН 10,0). В клетках, где рН среды близок к нейтральному, активность фермента очень низка.
Оксидаза D-аминокислот также обнаружена в почках и печени. Это FAD-зависимый фермент. Оптимум рН этой оксидазы лежит в нейтральной среде, поэтому фермент более активен, чем оксидаза L-аминокислот. Роль оксидазы D-аминокислот невелика, так как количество D-изомеров в организме крайне мало, потому что в белки пищи и белки тканей человека и животных входят только природные L-аминокислоты. Вероятно, оксидаза D-аминокислот способствует их превращению в соответствующие L-изомеры.
Большинство
аминокислот подвергается в клетке
непрямому
дезаминированию,
которое включает 2 стадии:
Трансаминирование с α-кетоглутаратом, образование Глу в цитозоле клетки
Окислительное дезаминирование Глу в митохондриях
Центральную роль в непрямом дезаминировании играют глутамат и α-кетоглутарат
Непрямое дезаминирование аминокислот происходит при участии 2 ферментов: аминотрансферазы (кофермент ПФ) и глутаматдегидрогеназы (кофермент NAD+).